
Nuvigil: Traitement de somnolence excessive (Information complète sur la prescription)
Nom de marque: Nuvigil
Nom générique: armodafinil
Comprimés Nuvigil® (armodafinil) [C-IV]
L'armodafinil est un médicament qui favorise l'éveil et qui est disponible sous forme de Nuvigil utilisé pour traiter l'apnée du sommeil, la narcolepsie ou les troubles du sommeil au travail posté. Utilisation, posologie, effets secondaires.
Contenu:
La description
Pharmacologie clinique
Essais cliniques
Indications et usage
Contre-indications
Avertissements
Précautions
Effets indésirables
Abus et dépendance aux drogues
Surdosage
Dosage et administration
Comment fournie
Fiche d'information patient Nuvigil (en anglais simple)
La description
NUVIGIL® (armodafinil) est un agent favorisant l'éveil pour l'administration orale. L'armodafinil est l'énantiomère R du modafinil qui est un mélange des énantiomères R et S. Le nom chimique de l'armodafinil est 2 - [(R) - (diphénylméthyl) sulfinyl] acétamide. La formule moléculaire est C15H15NON2S et le poids moléculaire est 273,35.
La structure chimique est:
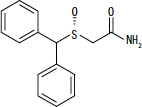
L'armodafinil est une poudre cristalline blanche à blanc cassé qui est très légèrement soluble dans l'eau, peu soluble dans l'acétone et soluble dans le méthanol. Les comprimés NUVIGIL contiennent 50, 150 ou 250 mg d'armodafinil et les ingrédients inactifs suivants: croscarmellose sodium, lactose monohydraté, stéarate de magnésium, cellulose microcristalline, povidone et prégélatinisé amidon.
Haut
Pharmacologie clinique
Mécanisme d'action et pharmacologie
Le ou les mécanismes précis par lesquels l'armodafinil (énantiomère R) ou le modafinil (mélange d'énantiomères R et S) favorisent l'éveil sont inconnus. L'armodafinil et le modafinil ont montré des propriétés pharmacologiques similaires dans des études non cliniques sur des animaux et in vitro, dans la mesure testée.
continuer l'histoire ci-dessous
Aux concentrations pharmacologiquement pertinentes, l'armodafinil ne se lie pas à ou n'inhibe pas plusieurs récepteurs et enzymes potentiellement pertinents pour régulation du sommeil / éveil, y compris celles pour la sérotonine, la dopamine, l'adénosine, la galanine, la mélatonine, la mélanocortine, l'orexine-1, l'orpheline, le PACAP ou benzodiazépines ou transporteurs du GABA, de la sérotonine, de la noradrénaline et de la choline ou de la phosphodiestérase VI, COMT, GABA transaminase et tyrosine hydroxylase. Le modafinil n'inhibe pas l'activité de la MAO-B ni des phosphodiestérases II-IV.
L'éveil induit par le modafinil peut être atténué par l'antagoniste des récepteurs α-1-adrénergiques, la prazosine; cependant, le modafinil est inactif dans d'autres systèmes de dosage in vitro connus pour être sensibles aux agonistes α-adrénergiques tels que la préparation du canal déférent du rat.
L'armodafinil n'est pas un agoniste des récepteurs de la dopamine à action directe ou indirecte. Cependant, in vitro, l'armodafinil et le modafinil se lient au transporteur de dopamine et inhibent la recapture de la dopamine. Pour le modafinil, cette activité a été associée in vivo à une augmentation des niveaux de dopamine extracellulaire dans certaines régions cérébrales des animaux. Chez les souris génétiquement modifiées dépourvues de transporteur de dopamine (DAT), le modafinil manquait d'activité favorisant le réveil, ce qui suggère que cette activité était dépendante du DAT. Cependant, les effets favorisant le réveil du modafinil, contrairement à ceux de l'amphétamine, n'ont pas été antagonisés par l'halopéridol, un antagoniste des récepteurs de la dopamine chez le rat.
De plus, l'alpha-méthyl-p-tyrosine, un inhibiteur de la synthèse de la dopamine, bloque l'action de l'amphétamine, mais ne bloque pas l'activité locomotrice induite par le modafinil.
L'armodafinil et le modafinil ont des actions favorisant le réveil similaires aux agents sympathomimétiques, y compris l'amphétamine et le méthylphénidate, bien que leur profil pharmacologique ne soit pas identique à celui du sympathomimétique amines. En plus de ses effets favorisant le réveil et de sa capacité à augmenter l'activité locomotrice chez les animaux, le modafinil produit effets psychoactifs et euphoriques, altérations de l'humeur, de la perception, de la pensée et des sentiments typiques des autres stimulants du SNC chez l'homme. Le modafinil a des propriétés renforçantes, comme en témoigne son auto-administration chez des singes préalablement formés à l'auto-administration de cocaïne; le modafinil a également été partiellement distingué comme un stimulant.
D'après des études non cliniques, deux métabolites majeurs, l'acide et la sulfone, du modafinil ou de l'armodafinil, ne semblent pas contribuer aux propriétés d'activation du SNC des composés parents.
Pharmacocinétique
Le composant actif de NUVIGIL est l'armodafinil, qui est l'énantiomère à vie plus longue du modafinil. NUVIGIL présente une cinétique linéaire indépendante du temps après l'administration d'une dose orale unique et multiple. L'augmentation de l'exposition systémique est proportionnelle sur la plage de doses de 50 à 400 mg. Aucun changement cinétique dépendant du temps n'a été observé au cours des 12 semaines de traitement. L'état d'équilibre apparent de NUVIGIL a été atteint dans les 7 jours suivant l'administration. À l'état d'équilibre, l'exposition systémique à NUVIGIL est 1,8 fois l'exposition observée après une dose unique. Les profils concentration-temps de l'énantiomère R pur après administration de 50 mg de NUVIGIL ou 100 mg de PROVIGIL® (modafinil) sont presque superposables.
Absorption
NUVIGIL est facilement absorbé après administration orale. La biodisponibilité orale absolue n'a pas été déterminée en raison de l'insolubilité aqueuse de l'armodafinil, qui a empêché l'administration intraveineuse. Les concentrations plasmatiques maximales sont atteintes à environ 2 heures à jeun. L'effet des aliments sur la biodisponibilité globale de NUVIGIL est considéré comme minime; cependant, le temps pour atteindre la concentration maximale (tmax) peut être retardé d'environ 2 à 4 heures à l'état alimenté. Depuis le retard de tmax est également associée à des taux plasmatiques élevés plus tard dans le temps, les aliments peuvent potentiellement affecter le début et le déroulement de l'action pharmacologique de NUVIGIL.
Distribution
NUVIGIL a un volume de distribution apparent d'environ 42 L. Les données spécifiques à la liaison aux protéines d'armodafinil ne sont pas disponibles. Cependant, le modafinil est modérément lié aux protéines plasmatiques (environ 60%), principalement à l'albumine. Le potentiel d'interactions de NUVIGIL avec des médicaments fortement liés aux protéines est considéré comme minime.
Métabolisme
Les données in vitro et in vivo montrent que l'armodafinil subit une désamidation hydrolytique, une S-oxydation et une hydroxylation du cycle aromatique, avec une conjugaison ultérieure de glucuronide des produits hydroxylés. L'hydrolyse des amides est la voie métabolique la plus importante, la formation de sulfones par le cytochrome P450 (CYP) 3A4 / 5 étant la prochaine importance. Les autres produits oxydants se forment trop lentement in vitro pour permettre l'identification de l'enzyme ou des enzymes responsables. Seuls deux métabolites atteignent des concentrations appréciables dans le plasma (c'est-à-dire l'acide R-modafinil et le modafinil sulfone).
Les données spécifiques à la disposition de NUVIGIL ne sont pas disponibles. Cependant, le modafinil est principalement éliminé par métabolisme, principalement dans le foie, avec moins de 10% du composé d'origine excrété dans l'urine. Au total, 81% de la radioactivité administrée a été récupérée dans les 11 jours suivant l'administration, principalement dans les urines (80% contre 1,0% dans les fèces).
Élimination
Après administration orale de NUVIGIL, l'armodafinil présente un déclin monoexponentiel apparent par rapport à la concentration plasmatique maximale. Le t ½ terminal apparent est d'environ 15 heures. La clairance orale de NUVIGIL est d'environ 33 ml / min.
Interactions médicament-médicament
L'existence de voies multiples pour le métabolisme de l'armodafinil, ainsi que le fait qu'une voie non liée au CYP est la plus rapide à métaboliser l'armodafinil, suggèrent qu'il existe une faible probabilité d'effets substantiels sur le profil pharmacocinétique global de NUVIGIL en raison de l'inhibition du CYP par médicaments.
Les données in vitro ont montré que l'armodafinil présente une faible réponse inductive pour le CYP1A2 et peut-être le CYP3A activités liées à la concentration et que l'activité du CYP2C19 est inhibée de manière réversible par armodafinil. Les autres activités du CYP ne semblent pas être affectées par l'armodafinil. Une étude in vitro a démontré que l'armodafinil est un substrat de la glycoprotéine P.
L'administration chronique de NUVIGIL à 250 mg a réduit l'exposition systémique au midazolam de 32% et 17% après administration orale unique (5 mg) et intraveineuse (2 mg), respectivement, ce qui suggère que l'administration de NUVIGIL induit modérément le CYP3A activité. Les médicaments qui sont des substrats du CYP3A4 / 5, comme la cyclosporine, peuvent nécessiter un ajustement posologique. (Voir Précautions, Interactions médicamenteuses).
L'administration chronique de NUVIGIL à 250 mg n'a pas affecté la pharmacocinétique de la caféine (200 mg), un substrat de sonde pour l'activité du CYP1A2.
L'administration concomitante d'une dose unique de 400 mg de NUVIGIL avec l'oméprazole (40 mg) a augmenté exposition à l'oméprazole d'environ 40%, ce qui indique que l'armodafinil inhibe modérément le CYP2C19 activité. Les médicaments qui sont des substrats du CYP2C19 peuvent nécessiter une réduction de la posologie. (Voir Précautions, Interactions médicamenteuses).
Effet de genre
L'analyse pharmacocinétique de population ne suggère aucun effet de genre sur la pharmacocinétique de l'armodafinil.
Populations spéciales
Les données spécifiques à l'armodafinil dans des populations particulières ne sont pas disponibles.
Effet de l'âge: Une légère diminution (~ 20%) de la clairance orale (CL / F) du modafinil a été observée dans une étude à dose unique à 200 mg sur 12 sujets avec un âge moyen de 63 ans (extrêmes 53 - 72 ans), mais le changement a été considéré comme non susceptible d'être cliniquement important. Dans une étude à doses multiples (300 mg / jour) chez 12 patients avec un âge moyen de 82 ans (intervalle de 67 à 87 ans), la moyenne les niveaux de modafinil dans le plasma étaient environ deux fois plus élevés que ceux historiquement sujets. En raison des effets potentiels des multiples médicaments concomitants avec lesquels la plupart des patients étaient traités, la différence apparente de pharmacocinétique du modafinil peut ne pas être uniquement attribuable aux effets vieillissement. Cependant, les résultats suggèrent que la clairance du modafinil peut être réduite chez les personnes âgées (voir Dosage et administration).
Effet de course: L'influence de la race sur la pharmacocinétique du modafinil n'a pas été étudiée.
Insuffisance rénale: Dans une étude à dose unique de 200 mg de modafinil, une insuffisance rénale chronique sévère (clairance de la créatinine - 20 ml / min) n'a pas influencer de façon significative la pharmacocinétique du modafinil, mais l'exposition à l'acide modafinil a été multipliée par 9 (voir Précautions).
Insuffisance hépatique: La pharmacocinétique et le métabolisme du modafinil ont été examinés chez des patients atteints de cirrhose du foie (6 hommes et 3 femmes). Trois patients avaient une cirrhose de stade B ou B + et 6 patients avaient une cirrhose de stade C ou C + (selon les critères de score de Child-Pugh). Cliniquement, 8 des 9 patients étaient ictériques et tous avaient une ascite. Chez ces patients, la clairance orale du modafinil a diminué d'environ 60% et la concentration à l'état d'équilibre a été doublée par rapport aux patients normaux. La dose de NUVIGIL doit être réduite chez les patients présentant une insuffisance hépatique sévère (voir Précautions et Dosage et administration).
Haut
Essais cliniques
L'efficacité de NUVIGIL dans l'amélioration de l'éveil a été établie dans le sommeil suivant troubles: syndrome d'apnée / hypopnée obstructive du sommeil (SAHOS), narcolepsie et troubles du sommeil par travail posté (SWSD).
Pour chaque essai clinique, une valeur de p de 0,05 € était requise pour la signification statistique.
Syndrome d'apnée / hypopnée obstructive du sommeil (SAHOS)
L'efficacité de NUVIGIL dans l'amélioration de l'éveil chez les patients présentant une somnolence excessive associée au SAHOS a été établie dans deux études multicentriques de 12 semaines, multicentriques, contrôlées contre placebo, en double aveugle, menées auprès de patients externes Critères de classification des troubles du sommeil (ICSD) pour le SAHOS (qui sont également conformes à l'American Psychiatric Association Critères DSM-IV). Ces critères comprennent: 1) une somnolence excessive ou une insomnie, ainsi que des épisodes fréquents de respiration pendant le sommeil et caractéristiques associées telles que ronflements bruyants, maux de tête le matin ou bouche sèche éveil; ou 2) somnolence excessive ou insomnie; et une polysomnographie montrant l'une des caractéristiques suivantes: plus de cinq apnées obstructives, chacune d'une durée supérieure à 10 secondes, par heure de sommeil; et un ou plusieurs des éléments suivants: réveils fréquents du sommeil associés aux apnées, bradytachycardie ou désaturation artérielle en oxygène en association avec les apnées. De plus, pour entrer dans ces études, tous les patients devaient avoir une somnolence excessive comme démontré par un score de 10 ¥ sur l'échelle de somnolence d'Epworth, malgré un traitement avec une pression positive continue (CPAP). Des preuves que la PPC était efficace pour réduire les épisodes d'apnée / hypopnée étaient nécessaires, ainsi qu'une documentation sur l'utilisation de la PPC.
Les patients devaient être conformes à la CPAP, définie comme une utilisation de la CPAP - ¥ 4 heures / nuit sur ‰ ¥ 70% des nuits. L'utilisation de CPAP s'est poursuivie tout au long de l'étude. Dans les deux études, les principales mesures de l'efficacité étaient 1) la latence du sommeil, telle qu'évaluée par le test de maintien de l'éveil (MWT) et 2) le changement de l'état général de la maladie du patient, tel que mesuré par l'impression clinique globale du changement (CGI-C) à la fin visite. Pour un essai réussi, les deux mesures devaient montrer une amélioration statistiquement significative.
Le MWT mesure la latence (en minutes) avant le début du sommeil. Un MWT étendu a été effectué avec des sessions de test à des intervalles de 2 heures entre 9 h et 19 h. L'analyse principale était la moyenne des latences de sommeil des quatre premières sessions de test (de 9 h à 15 h). Pour chaque session de test, le sujet a été invité à essayer de rester éveillé sans utiliser de mesures extraordinaires. Chaque session de test a été interrompue après 30 minutes en l'absence de sommeil ou immédiatement après le début du sommeil. Le CGI-C est une échelle à 7 points, centrée sur aucun changement, et allant de très pire à très amélioré. Les évaluateurs n'ont reçu aucune indication précise sur les critères à appliquer lors de l'évaluation des patients.
Dans la première étude, un total de 395 patients atteints de SAHOS ont été randomisés pour recevoir NUVIGIL 150 mg / jour, NUVIGIL 250 mg / jour ou un placebo correspondant. Les patients traités par NUVIGIL ont montré une amélioration statistiquement significative de la capacité de rester éveillé par rapport aux patients sous placebo, tel que mesuré par le MWT lors de la visite finale. Un plus grand nombre de patients traités statistiquement par NUVIGIL a montré une amélioration de l'état clinique global tel que évalué par l'échelle CGI-C lors de la visite finale. Les latences de sommeil moyennes (en minutes) dans le MWT au départ pour les essais sont présentées dans le tableau 1 ci-dessous, ainsi que la variation moyenne par rapport au départ sur le MWT lors de la visite finale. Les pourcentages de patients qui ont montré un quelconque degré d'amélioration du CGI-C dans les essais cliniques sont présentés dans le tableau 2 ci-dessous. Les deux doses de NUVIGIL ont produit des effets statistiquement significatifs d'amplitudes similaires sur le MWT, ainsi que sur le CGI-C.
Dans la deuxième étude, 263 patients atteints de SAHOS ont été randomisés pour recevoir NUVIGIL 150 mg / jour ou un placebo. Les patients traités par NUVIGIL ont montré une amélioration statistiquement significative de la capacité de rester éveillé par rapport aux patients sous placebo, tel que mesuré par le MWT [Tableau 1]. Un nombre statistiquement plus important de patients traités par NUVIGIL a montré une amélioration de l'état clinique global tel que évalué par l'échelle CGI-C [Tableau 2].
Le sommeil nocturne mesuré par polysomnographie n'a pas été affecté par l'utilisation de NUVIGIL dans les deux études.
Narcolepsie
L'efficacité de NUVIGIL dans l'amélioration de l'éveil chez les patients présentant une somnolence excessive (ES) associée à la narcolepsie a été établie dans une étude multicentrique, en double aveugle, multicentrique, contrôlée contre placebo, d'une durée de 12 semaines, menée auprès de patients ambulatoires répondant aux critères de l'ICSD narcolepsie. Au total, 196 patients ont été randomisés pour recevoir NUVIGIL 150 ou 250 mg / jour, ou un placebo correspondant. Les critères de l'ICSD pour la narcolepsie comprennent 1) des siestes récurrentes pendant la journée ou des interruptions de sommeil qui se produisent presque quotidiennement pendant au moins mois, plus une perte soudaine bilatérale de tonus musculaire postural en association avec une émotion intense (cataplexie), ou 2) une plainte de somnolence ou faiblesse musculaire soudaine avec des caractéristiques associées: paralysie du sommeil, hallucinations hypnagogiques, comportements automatiques, troubles majeurs épisode de sommeil; et une polysomnographie démontrant l'un des éléments suivants: latence du sommeil inférieure à 10 minutes ou latence du sommeil à mouvements oculaires rapides (REM) inférieure à 20 minutes et sommeil multiple Test de latence (MSLT) qui démontre une latence moyenne du sommeil de moins de 5 minutes et deux ou plusieurs périodes REM de début de sommeil et aucun trouble médical ou mental ne symptômes. Pour entrer dans ces études, tous les patients devaient avoir une somnolence diurne excessive documentée objectivement, via MSLT avec une latence de sommeil de 6 minutes ou moins et l'absence de toute autre activité médicale ou psychiatrique active cliniquement significative désordre. Le MSLT, une évaluation polysomnographique objective de la capacité du patient à s'endormir dans un environnement non stimulant, latence mesurée (en minutes) au début du sommeil en moyenne sur 4 sessions de test à Intervalles de 2 heures. Pour chaque session de test, on a dit au sujet de mentir tranquillement et d'essayer de dormir. Chaque session de test a été interrompue après 20 minutes si aucun sommeil ne s'était produit ou immédiatement après le début du sommeil.
Les principales mesures de l'efficacité étaient: 1) la latence du sommeil évaluée par le test de maintien de l'éveil (MWT) et 2) le changement de la maladie globale du patient état, tel que mesuré par l'impression clinique globale du changement (CGI-C) lors de la visite finale (voir la section ESSAIS CLINIQUES, SAHOS ci-dessus pour une description de ces les mesures). Chaque session de test MWT a été interrompue après 20 minutes si aucun sommeil ne s'est produit ou immédiatement après le début du sommeil dans cette étude.
Les patients traités par NUVIGIL ont montré une capacité statistiquement significativement améliorée de rester éveillé sur le MWT à chaque dose par rapport au placebo lors de la visite finale [Tableau 1]. Un nombre statistiquement plus important de patients traités par NUVIGIL à chaque dose a montré une amélioration de l'état clinique global tel que évalué par l'échelle CGI-C lors de la visite finale [Tableau 2].
Les deux doses de NUVIGIL ont produit des effets statistiquement significatifs d'amplitudes similaires sur le CGI-C. Bien qu'un effet statistiquement significatif sur le MWT ait été observé pour chaque dose, l'ampleur de l'effet a été observée plus grande pour la dose plus élevée.
Le sommeil nocturne mesuré par polysomnographie n'a pas été affecté par l'utilisation de NUVIGIL.
Trouble du sommeil au travail posté (SWSD)
L'efficacité de NUVIGIL dans l'amélioration de l'éveil chez les patients présentant une somnolence excessive associée à SWSD a été démontré dans une clinique multicentrique de 12 semaines, multicentrique, en double aveugle, contrôlée procès. Un total de 254 patients atteints de SWSD chronique ont été randomisés pour recevoir NUVIGIL 150 mg / jour ou un placebo. Tous les patients répondaient aux critères ICSD pour SWSD chronique [qui sont conformes aux critères DSM-IV de l'American Psychiatric Association pour le trouble du sommeil du rythme circadien: type de travail posté]. Ces critères comprennent 1) soit: a) une plainte principale de somnolence excessive ou d'insomnie qui est temporairement associée à une période de travail (généralement le travail de nuit) qui se produit pendant la phase de sommeil habituelle, ou b) la polysomnographie et le MSLT démontre la perte d'un schéma veille-sommeil normal (c.-à-d. perturbation chronobiologique rythmicité); et 2) aucun autre trouble médical ou mental ne explique les symptômes, et 3) les symptômes ne répondent pas aux critères pour tout autre trouble du sommeil entraînant une insomnie ou une somnolence excessive (par exemple, changement de fuseau horaire [décalage horaire] syndrome).
Il convient de noter que tous les patients présentant une plainte de somnolence qui sont également engagés dans un travail posté répondent aux critères de diagnostic de la SWSD. Dans l'essai clinique, seuls les patients présentant des symptômes pendant au moins 3 mois ont été inscrits.
Les patients inscrits devaient également travailler au moins 5 quarts de nuit par mois, avoir une somnolence excessive au l'heure de leurs quarts de nuit (score MSLT â ‰ ¤ 6 minutes), et ont une insomnie diurne documentée par un polysomnogramme diurne (PSG).
Les principales mesures d'efficacité étaient 1) la latence du sommeil, telle qu'évaluée par le test de latence du sommeil multiple (MSLT) effectué pendant un quart de nuit simulé à la visite finale, et 2) le changement de l'état général de la maladie du patient, tel que mesuré par l'impression clinique globale du changement (CGI-C) lors de la visite finale. (Voir Essais cliniques, Narcolepsie et OSAHS ci-dessus pour une description de ces mesures).
Les patients traités par NUVIGIL ont montré une prolongation statistiquement significative du temps de sommeil apparition par rapport aux patients sous placebo, telle que mesurée par le TMSL nocturne lors de la visite finale [Tableau 1]. Un nombre statistiquement plus élevé de patients traités par NUVIGIL a montré une amélioration de l'état clinique global tel que évalué par l'échelle CGI-C lors de la visite finale [Tableau 2].
Le sommeil diurne mesuré par polysomnographie n'a pas été affecté par l'utilisation de NUVIGIL.
| Désordre | Mesure | Nuvigil 150 mg * |
Nuvigil 250 mg * |
Placebo | |||
* Significativement différent du placebo pour tous les essais (p <0,05) | |||||||
| Référence | Changement de Baseline |
Référence | Changement de Baseline |
Référence | Changer de Référence |
||
| OSAHS I | MWT | 21.5 | 1.7 | 23.3 | 2.2 | 23.2 | -1.7 |
| OSAHS II | MWT | 23.7 | 2.3 | - | - | 23.3 | -1.3 |
| Narcolepsie | MWT | 12.1 | 1.3 | 9.5 | 2.6 | 12.5 | -1.9 |
| SWSD | MSLT | 2.3 | 3.1 | - | - | 2.4 | 0.4 |
| Désordre | Nuvigil 150 mg * |
Nuvigil 250 mg * |
Placebo |
* Significativement différent du placebo pour tous les essais (p <0,05) | |||
| OSAHS I | 71% | 74% | 37% |
| OSAHS II | 71% | - | 53% |
| Narcolepsie | 69% | 73% | 33% |
| SWSD | 79% | - | 59% |
Haut
Indications et usage
NUVIGIL est indiqué pour améliorer l'éveil chez les patients présentant une somnolence excessive associée au syndrome d'apnée / hypopnée obstructive du sommeil, à la narcolepsie et aux troubles du sommeil par travail posté.
Dans le SAHOS, NUVIGIL est indiqué en complément du (des) traitement (s) standard pour l'obstruction sous-jacente. Si la pression positive continue (CPAP) est le traitement de choix pour un patient, un effort maximal pour traiter avec CPAP pendant une période de temps adéquate doit être fait avant d'initier NUVIGIL. Si NUVIGIL est utilisé en association avec la PPC, l'encouragement et l'évaluation périodique de la conformité à la PPC sont nécessaires.
Dans tous les cas, une attention particulière au diagnostic et au traitement des troubles du sommeil sous-jacents est de la plus haute importance. Les prescripteurs doivent savoir que certains patients peuvent avoir plus d'un trouble du sommeil contribuant à leur somnolence excessive.
L'efficacité de NUVIGIL en utilisation à long terme (supérieure à 12 semaines) n'a pas été systématiquement évaluée dans les essais contrôlés versus placebo. Le médecin qui choisit de prescrire NUVIGIL pour une période prolongée chez les patients doit périodiquement réévaluer l'utilité à long terme pour le patient individuel.
Haut
Contre-indications
NUVIGIL est contre-indiqué chez les patients présentant une hypersensibilité connue au modafinil et à l'armodafinil ou à ses ingrédients inactifs.
Haut
AVERTISSEMENTS
Éruption cutanée grave, y compris le syndrome de Stevens-Johnson
Une éruption cutanée grave nécessitant une hospitalisation et l'arrêt du traitement a été rapportée chez des adultes en association avec l'armodafinil et chez l'adulte et l'enfant en association avec l'utilisation du modafinil, un mélange racémique de modafinil S et R (ce dernier est armodafinil).
L'armodafinil n'a été étudié chez les patients pédiatriques dans aucun contexte et son utilisation n'est approuvée chez les patients pédiatriques pour aucune indication.
Aucune éruption cutanée grave n'a été signalée dans les essais cliniques chez l'adulte (0 pour 1 595) d'armodafinil. Cependant, des cas d'éruptions cutanées graves ont été signalés chez des adultes au cours de l'expérience post-commercialisation. Étant donné que l'armodafinil est l'isomère R du modafinil racémique, un risque similaire d'éruption cutanée grave chez les patients pédiatriques sous armodafinil ne peut être exclu.
Dans les essais cliniques sur le modafinil (le racémate), l'incidence des éruptions cutanées entraînant l'arrêt du traitement était d'environ 0,8% (13 pour 1 585) chez les patients pédiatriques (âge <17 ans); ces éruptions cutanées comprenaient 1 cas de syndrome de Stevens-Johnson (SJS) possible et 1 cas de réaction d'hypersensibilité multi-organes apparente. Plusieurs des cas étaient associés à de la fièvre et à d'autres anomalies (par exemple, vomissements, leucopénie). Le délai médian d'éruption cutanée ayant entraîné l'arrêt du traitement était de 13 jours. Aucun cas de ce type n'a été observé chez 380 patients pédiatriques ayant reçu un placebo. Aucune éruption cutanée grave n'a été signalée dans les essais cliniques chez l'adulte (0 pour 4 264) de modafinil. Cas rares d'éruptions cutanées graves ou mettant la vie en danger, y compris SJS, nécrolyse épidermique toxique (RTE) et éruption cutanée médicamenteuse avec Une éosinophilie et des symptômes systémiques (DRESS) ont été signalés chez des adultes et des enfants dans le monde entier après la commercialisation. avec du modafinil. Le taux de déclaration des RTE et des SSJ associés à l'utilisation du modafinil, qui est généralement considéré comme une sous-estimation en raison de la sous-déclaration, dépasse le taux d'incidence de base. Les estimations du taux d'incidence de base de ces réactions cutanées graves dans la population générale varient entre 1 et 2 cas par million de personnes-années.
Il n'existe aucun facteur connu pour prédire le risque d'apparition ou la gravité des éruptions cutanées associées à l'armodafinil ou au modafinil. Presque tous les cas d'éruptions cutanées graves associées à l'armodafinil ou au modafinil sont survenus dans les 1 à 5 semaines suivant le début du traitement. Cependant, des cas isolés ont été signalés après un traitement prolongé avec du modafinil (par exemple, 3 mois). En conséquence, la durée du traitement ne peut pas être invoquée comme un moyen de prédire le risque potentiel annoncé par la première apparition d'une éruption cutanée.
Bien que des éruptions cutanées bénignes se produisent également avec l'armodafinil, il n'est pas possible de prédire de manière fiable quelles éruptions se révéleront graves. En conséquence, l'armodafinil doit normalement être arrêté dès le premier signe d'éruption cutanée, sauf si l'éruption n'est clairement pas liée au médicament. L'arrêt du traitement peut ne pas empêcher une éruption cutanée de mettre la vie en danger ou de devenir définitivement invalidante ou défigurante.
Angioedème et réactions anaphylactoïdes
Un cas grave d'œdème de Quincke et un cas d'hypersensibilité (avec éruption cutanée, dysphagie et bronchospasme) ont été observés chez 1 595 patients traités par l'armodafinil. Il faut conseiller aux patients d'arrêter le traitement et de signaler immédiatement à leur médecin tout signe ou symptômes suggérant un œdème de Quincke ou une anaphylaxie (par exemple, gonflement du visage, des yeux, des lèvres, de la langue ou larynx; difficulté à avaler ou à respirer; enrouement).
Réactions d'hypersensibilité multi-organes
Les réactions d'hypersensibilité multi-organes, y compris au moins un décès dans l'expérience post-commercialisation, ont survenue dans une association temporelle étroite (délai médian de détection de 13 jours: plage de 4 à 33) au début de modafinil. Un risque similaire de réactions d'hypersensibilité multi-organes avec l'armodafinil ne peut être exclu.
Bien qu'il y ait eu un nombre limité de rapports, les réactions d'hypersensibilité multi-organes peuvent entraîner une hospitalisation ou mettre la vie en danger. Il n'y a aucun facteur connu pour prédire le risque d'apparition ou la gravité des réactions d'hypersensibilité multi-organes associées au modafinil. Les signes et symptômes de ce trouble étaient divers; cependant, les patients présentaient généralement, mais pas exclusivement, de la fièvre et des éruptions cutanées associées à une autre atteinte du système organique. Les autres manifestations associées comprenaient la myocardite, l'hépatite, les anomalies des tests de la fonction hépatique, anomalies hématologiques (par exemple, éosinophilie, leucopénie, thrombocytopénie), prurit et asthénie. Parce que l'hypersensibilité multi-organes est variable dans son expression, d'autres symptômes et signes du système organique, non mentionnés ici, peuvent survenir.
Si une réaction d'hypersensibilité multi-organes est suspectée, NUVIGIL doit être arrêté. Bien qu'il n'y ait aucun rapport de cas indiquant une sensibilité croisée avec d'autres médicaments qui produisent ce syndrome, l'expérience avec des médicaments associés à une hypersensibilité multi-organes indiquerait qu'il s'agit d'un possibilité.
Somnolence persistante
Les patients présentant une somnolence anormale qui prennent NUVIGIL doivent être informés que leur niveau d'éveil peut ne pas revenir à la normale. Les patients présentant une somnolence excessive, y compris ceux qui prennent NUVIGIL, doivent être fréquemment réévalués pour degré de somnolence et, le cas échéant, conseillé d'éviter de conduire ou tout autre danger potentiel activité. Les prescripteurs doivent également être conscients que les patients peuvent ne pas reconnaître la somnolence ou la somnolence avant d'être directement interrogés sur la somnolence ou la somnolence lors d'activités spécifiques.
Symptômes psychiatriques
Des effets indésirables psychiatriques ont été rapportés chez des patients traités par le modafinil. Le modafinil et l'armodafinil (NUVIGIL) sont très étroitement liés. Par conséquent, l'incidence et le type de symptômes psychiatriques associés à l'armodafinil devraient être similaires à l'incidence et au type de ces événements avec le modafinil.
Les événements indésirables post-commercialisation associés à l'utilisation du modafinil ont inclus la manie, les délires, les hallucinations, les idées suicidaires et l'agression, certains entraînant une hospitalisation. De nombreux patients, mais pas tous, avaient des antécédents psychiatriques. Un volontaire masculin en bonne santé a développé des idées de référence, des délires paranoïdes et des hallucinations auditives en association avec de multiples doses quotidiennes de 600 mg de modafinil et une privation de sommeil. Il n'y avait aucun signe de psychose 36 heures après l'arrêt du médicament.
Dans la base de données de l'essai contrôlé NUVIGIL, l'anxiété, l'agitation, la nervosité et l'irritabilité étaient des raisons l'arrêt du traitement plus souvent chez les patients sous NUVIGIL par rapport au placebo (NUVIGIL 1,2% et placebo 0.3%). Dans les études contrôlées par NUVIGIL, la dépression était également une raison pour l'arrêt du traitement plus souvent chez les patients sous NUVIGIL par rapport au placebo (NUVIGIL 0,6% et placebo 0,2%). Deux cas d'idées de suicide ont été observés dans les essais cliniques. Il faut être prudent lorsque NUVIGIL est administré à des patients ayant des antécédents de psychose, de dépression ou de manie. Si des symptômes psychiatriques se développent en association avec l'administration de NUVIGIL, envisagez d'arrêter NUVIGIL.
Haut
PRÉCAUTIONS
Diagnostic des troubles du sommeil
NUVIGIL ne doit être utilisé que chez les patients qui ont subi une évaluation complète de leur somnolence excessive et chez le diagnostic de narcolepsie, SAHOS et / ou SWSD a été établi conformément aux critères de diagnostic ICSD ou DSM (voir Essais cliniques). Une telle évaluation consiste généralement en un historique complet et un examen physique, et elle peut être complétée par des tests en laboratoire. Certains patients peuvent avoir plus d'un trouble du sommeil contribuant à leur somnolence excessive (par exemple, OSAHS et SWSD coïncident chez le même patient).
Utilisation de CPAP chez les patients atteints de SAHOS
Dans le SAHOS, NUVIGIL est indiqué en complément du (des) traitement (s) standard pour l'obstruction sous-jacente. Si la pression positive continue (CPAP) est le traitement de choix pour un patient, un effort maximal pour traiter avec CPAP pendant une période de temps adéquate doit être fait avant d'initier NUVIGIL. Si NUVIGIL est utilisé en association avec la PPC, l'encouragement et l'évaluation périodique de la conformité à la PPC sont nécessaires. Il y avait une légère tendance à une réduction de l'utilisation de la CPAP au fil du temps (réduction moyenne de 18 minutes pour les patients traités par NUVIGIL et une réduction de 6 minutes pour les patients sous placebo à partir d'une utilisation initiale moyenne de 6,9 heures par nuit) dans NUVIGIL essais.
Général
Bien qu'il n'ait pas été démontré que NUVIGIL provoque une altération fonctionnelle, tout médicament affectant le SNC peut altérer le jugement, la réflexion ou la motricité. Les patients doivent être avertis de l'utilisation d'une automobile ou d'autres machines dangereuses jusqu'à ce raisonnablement certain que la thérapie NUVIGIL n'affectera pas négativement leur capacité à Activités.
Système cardiovasculaire
NUVIGIL n'a pas été évalué ou utilisé dans une mesure appréciable chez les patients ayant des antécédents récents d'infarctus du myocarde ou d'angor instable, et ces patients doivent être traités avec prudence.
Dans les études cliniques de PROVIGIL, signes et symptômes, y compris douleurs thoraciques, palpitations, dyspnée et ischémie transitoire Des modifications des ondes T sur l'ECG ont été observées chez trois sujets en association avec un prolapsus valvulaire mitral ou un ventricule gauche hypertrophie. Il est recommandé de ne pas utiliser les comprimés NUVIGIL chez les patients ayant des antécédents d'hypertrophie ventriculaire gauche ou les patients atteints de prolapsus valvulaire mitral qui ont éprouvé le syndrome de prolapsus valvulaire mitral lorsqu'ils recevaient auparavant un SNC stimulants. Les signes du syndrome du prolapsus valvulaire mitral incluent, sans s'y limiter, les changements d'ECG ischémiques, les douleurs thoraciques ou l'arythmie. En cas d'apparition d'un nouveau de ces symptômes, envisager une évaluation cardiaque.
La surveillance de la pression artérielle dans des essais contrôlés à court terme (3 mois) n'a montré que de légères augmentations moyennes des pression artérielle diastolique chez les patients recevant NUVIGIL par rapport au placebo (1,2 à 4,3 mmHg dans les groupes). Il y avait également une proportion légèrement plus élevée de patients sous NUVIGIL nécessitant une utilisation nouvelle ou accrue de médicaments antihypertenseurs (2,9%) par rapport aux patients sous placebo (1,8%). Une surveillance accrue de la pression artérielle peut être appropriée chez les patients sous NUVIGIL.
Patients utilisant des contraceptifs stéroïdiens
L'efficacité des contraceptifs stéroïdiens peut être réduite lorsqu'ils sont utilisés avec NUVIGIL et pendant un mois après l'arrêt du traitement (voir Précautions, Interactions médicamenteuses). Des méthodes de contraception alternatives ou concomitantes sont recommandées pour les patientes traitées par NUVIGIL et pendant un mois après l'arrêt du traitement par NUVIGIL.
Patients utilisant de la cyclosporine
Les taux sanguins de cyclosporine peuvent être réduits lorsqu'ils sont utilisés avec NUVIGIL (voir Précautions, Interactions médicamenteuses). La surveillance des concentrations de cyclosporine en circulation et un ajustement approprié de la posologie de la cyclosporine doivent être envisagés lorsque ces médicaments sont utilisés en concomitance.
Patients atteints d'insuffisance hépatique sévère
Chez les patients atteints d'insuffisance hépatique sévère, avec ou sans cirrhose (voir Pharmacologie clinique), NUVIGIL doit être administré à une dose réduite (voir Dosage et administration).
Patients atteints d'insuffisance rénale sévère
Les informations sont insuffisantes pour déterminer l'innocuité et l'efficacité du dosage chez les patients présentant une insuffisance rénale sévère (pour la pharmacocinétique de l'insuffisance rénale, voir Pharmacologie clinique).
Patients âgés
Chez les patients âgés, l'élimination de l'armodafinil et de ses métabolites peut être réduite en raison du vieillissement. Par conséquent, il convient d'envisager l'utilisation de doses plus faibles dans cette population (voir Pharmacologie clinique et Dosage et administration).
Information pour les patients
Il est conseillé aux médecins de discuter des problèmes suivants avec les patients auxquels ils prescrivent NUVIGIL.
NUVIGIL est indiqué pour les patients qui ont des niveaux anormaux de somnolence. Il a été démontré que NUVIGIL améliore, mais n'élimine pas, cette tendance anormale à s'endormir. Par conséquent, les patients ne doivent pas modifier leur comportement antérieur en ce qui concerne les activités potentiellement dangereuses (par exemple, conduire, utiliser des machines) ou d'autres les activités nécessitant des niveaux d'éveil appropriés, jusqu'à ce qu'il soit démontré que le traitement par NUVIGIL produit des niveaux d'éveil permettant une telle Activités. Les patients doivent être informés que NUVIGIL ne remplace pas le sommeil.
Les patients doivent être informés qu'il peut être essentiel qu'ils continuent de prendre leurs traitements précédemment prescrits (par exemple, les patients atteints de SAHOS recevant la PPC devraient continuer à le faire).
Les patients doivent être informés de la disponibilité d'une notice d'information pour les patients et doivent être informés de la lecture de la notice avant de prendre NUVIGIL. Voir les informations destinées aux patients à la fin de cet étiquetage pour le texte de la notice fournie aux patients.
Il faut conseiller aux patients de contacter leur médecin en cas d'éruption cutanée, de dépression, d'anxiété ou de signes de psychose ou de manie.
Grossesse
Il faut conseiller aux patientes d'aviser leur médecin si elles tombent enceintes ou ont l'intention de devenir enceintes pendant le traitement. Les patientes doivent être mises en garde concernant le risque potentiel accru de grossesse lors de l'utilisation de contraceptifs stéroïdiens (y compris les dépôts ou les implants implantables). contraceptifs) avec NUVIGIL et pendant un mois après l'arrêt du traitement (voir Carcinogenèse, Mutagenèse, Affaiblissement de la Fertilité et Grossesse).
Allaitement
On devrait conseiller aux patients d'informer leur médecin s'ils allaitent un bébé.
Médicament concomitant
Les patients doivent être informés d'informer leur médecin s'ils prennent ou envisagent de prendre tout médicaments en vente libre ou en vente libre, en raison du potentiel d'interactions entre NUVIGIL et d'autres drogues.
De l'alcool
Les patients doivent être informés que l'utilisation de NUVIGIL en association avec de l'alcool n'a pas été étudiée. Les patients doivent être informés qu'il est prudent d'éviter l'alcool lors de la prise de NUVIGIL.
Réactions allergiques
On devrait conseiller aux patients d'arrêter de prendre NUVIGIL et d'aviser leur médecin s'ils développent une éruption cutanée, urticaire, plaies dans la bouche, cloques, desquamation de la peau, difficulté à avaler ou à respirer ou un allergique apparenté phénomène.
Interactions médicamenteuses
Interactions potentielles avec des médicaments qui inhibent, induisent ou sont métabolisés par les isoenzymes du cytochrome P450 et d'autres enzymes hépatiques
En raison de l'implication partielle des enzymes CYP3A dans l'élimination métabolique de l'armodafinil, la co-administration d'inducteurs puissants du CYP3A4 / 5 (p. ex. carbamazépine, phénobarbital, rifampicine) ou des inhibiteurs du CYP3A4 / 5 (p. ex. kétoconazole, érythromycine) pourraient modifier les taux plasmatiques de armodafinil.
Le potentiel de NUVIGIL pour modifier le métabolisme d'autres médicaments par induction ou inhibition enzymatique
Médicaments métabolisés par le CYP1A2: Les données in vitro ont montré que l'armodafinil présente une faible réponse inductive pour le CYP1A2 et peut-être le CYP3A activités liées à la concentration et a démontré que l'activité du CYP2C19 est inhibée de façon réversible par armodafinil. Cependant, l'effet sur l'activité du CYP1A2 n'a pas été observé cliniquement dans une étude d'interaction réalisée avec de la caféine (voir Pharmacologie clinique, Pharmacocinétique, interactions médicamenteuses).
Médicaments métabolisés par le CYP3A4 / 5 (par exemple, la cyclosporine, l'éthinylestradiol, le midazolam et le triazolam): L'administration chronique de NUVIGIL a entraîné une induction modérée de l'activité du CYP3A. Par conséquent, l'efficacité des médicaments qui sont des substrats des enzymes CYP3A (par exemple, la cyclosporine, l'éthinyl œstradiol, midazolam et triazolam) peuvent être réduits après l'instauration d'un traitement concomitant par NUVIGIL. Une réduction de 32% de l'exposition systémique au midazolam par voie orale a été observée lors de l'administration concomitante d'armodafinil et de midazolam. Un ajustement de la dose peut être nécessaire (voir Pharmacologie clinique, Pharmacocinétique, interactions médicamenteuses). De tels effets (concentrations réduites) ont également été observés lors de l'administration concomitante de modafinil et de cyclosporine, d'éthinylœstradiol et de triazolam.
Médicaments métabolisés par le CYP2C19 (p. Ex., Oméprazole, diazépam, phénytoïne et propranolol): L'administration de NUVIGIL a entraîné une inhibition modérée de l'activité du CYP2C19. Par conséquent, une réduction de la dose peut être nécessaire pour certains médicaments qui sont des substrats du CYP2C19 (par exemple, la phénytoïne, le diazépam et le propranolol, l'oméprazole et la clomipramine) lorsqu'ils sont utilisés en même temps que NUVIGIL. Une augmentation de 40% de l'exposition a été observée lors de l'administration concomitante d'armodafinil et d'oméprazole. (Voir Pharmacologie clinique, Pharmacocinétique, interactions médicamenteuses).
Interactions avec le SNC AMédicaments actifs
Les données spécifiques au potentiel d'interaction médicament-armodafinil avec les médicaments actifs sur le SNC ne sont pas disponibles. Cependant, les informations d'interaction médicamenteuse disponibles sur le modafinil suivantes devraient être applicables à l'armodafinil (voir La description et Pharmacologie clinique).
L'administration concomitante de modafinil et de méthylphénidate ou de dextroamphétamine n'a produit aucune altération significative profil pharmacocinétique du modafinil ou de l'un ou l'autre des stimulants, même si l'absorption du modafinil a été retardée d'environ un heure.
Le modafinil ou la clomipramine concomitants n'ont pas modifié le profil pharmacocinétique de l'un ou l'autre médicament; cependant, un incident d'augmentation des niveaux de clomipramine et de son métabolite actif, la desméthylclomipramine, a été signalé chez un patient atteint de narcolepsie pendant le traitement par le modafinil.
Les données spécifiques au potentiel d'interaction médicamenteuse de l'armodafinil ou du modafinil avec les inhibiteurs de la monoamine oxydase (MAO) ne sont pas disponibles. Par conséquent, la prudence s'impose lors de l'administration concomitante d'inhibiteurs de la MAO et de NUVIGIL.
Interactions avec d'autres médicaments
Les données spécifiques au potentiel d'interaction médicament-armodafinil pour d'autres médicaments supplémentaires ne sont pas disponibles. Cependant, les informations disponibles sur l'interaction médicamenteuse disponibles sur le modafinil devraient être applicables à l'armodafinil.
Warfarine - L'administration concomitante de modafinil et de warfarine n'a pas entraîné de modifications significatives des profils pharmacocinétiques de la R et de la S-warfarine. Cependant, comme une seule dose de warfarine a été testée dans cette étude, une interaction pharmacodynamique ne peut être exclue. Par conséquent, une surveillance plus fréquente des temps de prothrombine / INR doit être envisagée chaque fois que NUVIGIL est co-administré avec la warfarine.
Carcinogenèse, mutagenèse, altération de la fertilité
Cancérogenèse
Aucune étude de cancérogénicité n'a été menée avec l'armodafinil seul. Des études de cancérogénicité ont été menées dans lesquelles du modafinil a été administré dans l'alimentation à des souris pendant 78 semaines et à des rats pendant 104 semaines à des doses de 6, 30 et 60 mg / kg / jour. La dose la plus élevée étudiée représente 1,5 (souris) ou 3 (rat) fois supérieure à la dose quotidienne recommandée de modafinil chez l'homme adulte (200 mg) en mg / m2. Il n'y avait aucune preuve de tumorigenèse associée à l'administration de modafinil dans ces études. Cependant, étant donné que l'étude sur la souris a utilisé une dose élevée inadéquate qui n'était pas représentative d'une dose maximale tolérée, une étude ultérieure de cancérogénicité a été menée dans la Tg. Souris transgénique AC. Doses évaluées dans la Tg. Le dosage AC était de 125, 250 et 500 mg / kg / jour, administré par voie cutanée. Il n'y avait aucune preuve de tumorigénicité associée à l'administration de modafinil; cependant, ce modèle cutané peut ne pas évaluer adéquatement le potentiel cancérogène d'un médicament administré par voie orale.
Mutagenèse
L'armodafinil a été évalué dans un essai de mutation inverse bactérienne in vitro et dans un essai d'aberration chromosomique de mammifère in vitro sur des lymphocytes humains. L'armodafinil s'est révélé négatif dans ces tests, tant en l'absence qu'en présence d'activation métabolique.
Le modafinil n'a démontré aucun signe de potentiel mutagène ou clastogène dans une série de tests in vitro (c.-à-d., Test de mutation inverse bactérienne, test tk de lymphome de souris, aberration chromosomique test dans les lymphocytes humains, test de transformation cellulaire dans des cellules d'embryon de souris BALB / 3T3) dosages en l'absence ou en présence d'activation métabolique, ou in vivo (micronoyau de moelle osseuse de souris) dosages. Le modafinil était également négatif dans le test de synthèse d'ADN non programmé dans les hépatocytes de rat.
Atteinte à la fertilité
Aucune étude de fertilité et de développement embryonnaire précoce (jusqu'à l'implantation) n'a été menée avec l'armodafinil seul.
Administration orale de modafinil (doses allant jusqu'à 480 mg / kg / jour) à des rats mâles et femelles avant et pendant toute la durée l'accouplement et la poursuite des femelles jusqu'au 7e jour de gestation ont entraîné une augmentation du temps d'accouplement au plus haut dose; aucun effet n'a été observé sur d'autres paramètres de fertilité ou de reproduction. La dose sans effet de 240 mg / kg / jour a été associée à une exposition plasmatique au modafinil (ASC) approximativement égale à celle chez l'homme à la dose recommandée de 200 mg.
Grossesse
Catégorie de grossesse C.
Dans des études menées chez le rat (armodafinil, modafinil) et le lapin (modafinil), une toxicité pour le développement a été observée à des expositions cliniquement pertinentes.
L'administration orale d'armodafinil (60, 200 ou 600 mg / kg / jour) à des rates gravides tout au long de la période d'organogenèse a entraîné une augmentation l'incidence des variations viscérales et squelettiques fœtales à la dose intermédiaire ou à des poids corporels fœtaux plus élevés et plus faibles au plus haut dose. La dose sans effet pour la toxicité sur le développement embryofœtal du rat a été associée à un armodafinil plasmatique (ASC) environ 0,03 fois l'ASC chez l'homme à la dose quotidienne maximale recommandée de 250 mg.
Le modafinil (50, 100 ou 200 mg / kg / jour) administré par voie orale à des rates gravides pendant toute la période d’organogenèse a provoqué, en l’absence de toxicité maternelle, une augmentation des résorptions et une incidence accrue de variations viscérales et squelettiques chez la progéniture au plus haut dose. La dose sans effet plus élevée pour la toxicité pour le développement embryofœtal du rat a été associée à un exposition au modafinil environ 0,5 fois l'ASC chez l'homme à la dose quotidienne recommandée (RHD) de 200 mg. Cependant, dans une étude ultérieure allant jusqu'à 480 mg / kg / jour (exposition plasmatique au modafinil environ 2 fois l'ASC chez l'homme au RHD), aucun effet indésirable sur le développement embryofœtal n'a été observé.
Modafinil administré par voie orale à des lapines gravides tout au long de la période d'organogenèse à des doses allant jusqu'à 100 mg / kg / jour (ASC de modafinil plasmatique approximativement égale à l'ASC chez l'homme au RHD) n'a eu aucun effet sur l'embryo-fœtus développement; cependant, les doses utilisées étaient trop faibles pour évaluer adéquatement les effets du modafinil sur le développement embryofœtal. Dans une étude ultérieure de toxicité pour le développement évaluant des doses de 45, 90 et 180 mg / kg / jour chez les femmes enceintes lapins, l'incidence des altérations de la structure fœtale et de la mort embryofœtale a augmenté au plus haut dose. La dose sans effet la plus élevée pour la toxicité pour le développement a été associée à une ASC plasmatique du modafinil approximativement égale à l'ASC chez l'homme au RHD.
L'administration de modafinil à des rats pendant la gestation et la lactation à des doses orales allant jusqu'à 200 mg / kg / jour a entraîné une diminution la viabilité de la progéniture à des doses supérieures à 20 mg / kg / jour (ASC plasmatique du modafinil environ 0,1 fois l'ASC chez l'homme au RHD). Aucun effet sur les paramètres développementaux et neurocomportementaux postnatals n'a été observé chez les descendants survivants.
Il n'y a pas d'études adéquates et bien contrôlées sur l'armodafinil ou le modafinil chez les femmes enceintes. Deux cas de retard de croissance intra-utérin et un cas d'avortement spontané ont été signalés en association avec l'armodafinil et le modafinil. Bien que la pharmacologie de l'armodafinil ne soit pas identique à celle des amines sympathomimétiques, il partage certaines propriétés pharmacologiques avec cette classe. Certains de ces médicaments ont été associés à un retard de croissance intra-utérin et à des avortements spontanés. On ignore si les cas signalés avec l'armodafinil sont liés à la drogue.
L'armodafinil ou le modafinil ne doit être utilisé pendant la grossesse que si le bénéfice potentiel justifie le risque potentiel pour le fœtus.
Travail et accouchement
L'effet de l'armodafinil sur le travail et l'accouchement chez l'homme n'a pas été systématiquement étudié.
Mères allaitantes
On ne sait pas si l'armodafinil ou ses métabolites sont excrétés dans le lait maternel. Étant donné que de nombreux médicaments sont excrétés dans le lait maternel, la prudence s'impose lorsque les comprimés NUVIGIL sont administrés à une femme qui allaite.
Utilisation pédiatrique
La sécurité et l'efficacité de l'utilisation de l'armodafinil chez les personnes de moins de 17 ans n'ont pas été établies. Une éruption cutanée grave a été observée chez des patients pédiatriques recevant du modafinil
Utilisation géiratrique
La sécurité et l'efficacité chez les personnes de plus de 65 ans n'ont pas été établies.
Haut
Effets indésirables
Armodafinil a été évalué pour l'innocuité chez plus de 1100 patients présentant une somnolence excessive associée à des troubles primaires du sommeil et de l'éveil. Dans les essais cliniques, NUVIGIL s'est avéré généralement bien toléré et la plupart des effets indésirables ont été légers à modérés.
Dans les études cliniques contrôlées contre placebo, les événements indésirables les plus fréquemment observés (‰ ¥ 5%) associés à l'utilisation de NUVIGIL survenant plus fréquemment que chez les patients sous placebo étaient des céphalées, des nausées, des étourdissements et insomnie. Le profil des événements indésirables était similaire dans toutes les études.
Dans les essais cliniques contrôlés contre placebo, 44 des 645 patients (7%) qui ont reçu NUVIGIL ont abandonné en raison d'une expérience indésirable, contre 16 des 445 (4%) des patients qui ont reçu le placebo. La raison la plus fréquente d'arrêt était le mal de tête (1%).
Incidence dans les essais contrôlés
Le tableau suivant (tableau 3) présente les expériences indésirables survenues à un taux de 1% ou plus et plus fréquente chez les patients traités par NUVIGIL que chez les patients du groupe placebo dans les essais cliniques contrôlés versus placebo.
Le prescripteur doit savoir que les chiffres fournis ci-dessous ne peuvent pas être utilisés pour prédire la fréquence des événements indésirables cours de la pratique médicale habituelle, où les caractéristiques des patients et d'autres facteurs peuvent différer de ceux qui se études. De même, les fréquences citées ne peuvent pas être directement comparées aux chiffres obtenus à partir d'autres investigations cliniques impliquant différents traitements, utilisations ou investigateurs. Cependant, l'examen de ces fréquences fournit aux prescripteurs une base pour estimer la contribution relative des facteurs médicamenteux et non médicamenteux à l'incidence des événements indésirables dans la population étudiée.
| Classe System Organ Terme privilégié MedDRA |
Nuvigil (Pourcentage, N = 645) |
Placebo (Pourcentage, N = 445) |
ª Quatre études cliniques en double aveugle, contrôlées par placebo, sur le SWSD, le SAHOS et la narcolepsie; l'incidence est arrondie au pourcentage entier le plus proche. Sont inclus uniquement les événements pour lesquels l'incidence de Nuvigil est supérieure à celle du placebo. | ||
| Troubles cardiaques | ||
| Palpitations | 2 | 1 |
| Problèmes gastro-intestinaux | ||
| La nausée | 7 | 3 |
| La diarrhée | 4 | 2 |
| Bouche sèche | 4 | 1 |
| Dyspepsie | 2 | 0 |
| Douleur abdominale supérieure | 2 | 1 |
| Constipation | 1 | 0 |
| Vomissement | 1 | 0 |
| Selles molles | 1 | 0 |
| Troubles généraux et conditions du site d'administration | ||
| Fatigue | 2 | 1 |
| La soif | 1 | 0 |
| Maladie grippale | 1 | 0 |
| Douleur | 1 | 0 |
| Pyrexia | 1 | 0 |
| Troubles du système immunitaire | ||
| Allergie saisonnière | 1 | 0 |
| Enquêtes | ||
| Augmentation de la gamma-glutamyltransférase | 1 | 0 |
| Augmentation de la fréquence cardiaque | 1 | 0 |
| Troubles du métabolisme et de la nutrition | ||
| Anorexie | 1 | 0 |
| Diminution de l'appétit | 1 | 0 |
| Troubles du système nerveux | ||
| Mal de crâne | 17 | 9 |
| Vertiges | 5 | 2 |
| Perturbation en attention | 1 | 0 |
| Tremblement | 1 | 0 |
| Migraine | 1 | 0 |
| Paresthésie | 1 | 0 |
| Troubles psychiatriques | ||
| Insomnie | 5 | 1 |
| Anxiété | 4 | 1 |
| La dépression | 2 | 0 |
| Agitation | 1 | 0 |
| Nervosité | 1 | 0 |
| Humeur dépressive | 1 | 0 |
| Troubles rénaux et urinaires | ||
| Polyurie | 1 | 0 |
| Troubles respiratoires, thoraciques et médiastinaux | ||
| Dyspnée | 1 | 0 |
| Troubles de la peau et des tissus sous-cutanés | ||
| Éruption | 2 | 0 |
| Dermatite de contact | 1 | 0 |
| Hyperhydrose | 1 | 0 |
Dépendance à la dose des événements indésirables
Dans les essais cliniques contrôlés contre placebo qui ont comparé des doses de 150 mg / jour et 250 mg / jour de Nuvigil et du placebo, les seuls événements indésirables qui semblaient être liés à la dose étaient les maux de tête, les éruptions cutanées, la dépression, la bouche sèche, l'insomnie et la nausée.
ª Quatre études cliniques en double aveugle, contrôlées contre placebo, sur le SWSD, le SAHOS et la narcolepsie. | ||||
| Classe System Organ Terme privilégié MedDRA |
Nuvigil 250 mg (Pour cent, N = 198) |
Nuvigil 150 mg (Pour cent, N = 447) |
Nuvigil Combiné (Pour cent, N = 645) |
Placebo (Pour cent, N = 445) |
|---|---|---|---|---|
| Problèmes gastro-intestinaux | ||||
| La nausée | 9 | 6 | 7 | 3 |
| Bouche sèche | 7 | 2 | 4 | <1 |
| Troubles du système nerveux | ||||
| Mal de crâne | 23 | 14 | 17 | 9 |
| Troubles psychiatriques | ||||
| Insomnie | 6 | 4 | 5 | 1 |
| La dépression | 3 | 1 | 2 | <1 |
| Troubles de la peau et des tissus sous-cutanés | ||||
| Éruption | 4 | 1 | 2 | <1 |
Modifications des signes vitaux
Il y a eu des augmentations faibles, mais constantes, des valeurs moyennes de la pression artérielle systolique et diastolique moyenne dans les essais contrôlés (voir Précautions). Il y a eu une augmentation moyenne faible, mais constante, du pouls par rapport au placebo dans les essais contrôlés. Cette augmentation variait de 0,9 à 3,5 BPM.
Changements de laboratoire
Les paramètres de chimie clinique, d'hématologie et d'analyse d'urine ont été surveillés dans les études. Les niveaux plasmatiques moyens de gamma glutamyltransférase (GGT) et de phosphatase alcaline (AP) se sont révélés plus élevés après l'administration de NUVIGIL, mais pas de placebo. Cependant, peu de sujets avaient des élévations de GGT ou AP en dehors de la plage normale. Aucune différence n’était apparente entre l’alanine aminotransférase, l’aspartate aminotransférase, la protéine totale, l'albumine ou la bilirubine totale, bien qu'il y ait eu de rares cas d'élévations isolées d'AST et / ou d'ALT. Un seul cas de pancytopénie légère a été observé après 35 jours de traitement et a disparu avec l'arrêt du médicament. Une légère diminution moyenne par rapport aux valeurs initiales de l'acide urique sérique par rapport au placebo a été observée dans les essais cliniques. La signification clinique de cette découverte est inconnue.
Modifications de l'ECG
Aucun schéma d'anomalies ECG n'a pu être attribué à l'administration de NUVIGIL dans les essais cliniques contrôlés versus placebo.
Haut
Abus et dépendance aux drogues
Classe de substances contrôlées
L'armodafinil (NUVIGIL) est une substance réglementée de l'annexe IV.
Potentiel d'abus et dépendance
Bien que le potentiel d'abus de l'armodafinil n'ait pas été spécifiquement étudié, son potentiel d'abus est susceptible d'être similaire à celui du modafinil (PROVIGIL). Chez l'homme, le modafinil produit des effets psychoactifs et euphoriques, des altérations de l'humeur, de la perception, de la pensée et des sentiments typiques des autres stimulants du SNC. Dans les études de liaison in vitro, le modafinil se lie au site de recapture de la dopamine et provoque une augmentation de la dopamine extracellulaire, mais aucune augmentation de la libération de dopamine. Le modafinil se renforce, comme en témoigne son auto-administration chez des singes préalablement formés à l'auto-administration de cocaïne. Dans certaines études, le modafinil a également été partiellement distingué comme un stimulant. Les médecins doivent suivre les patients de près, en particulier ceux qui ont des antécédents d'abus de drogues et / ou de stimulants (par exemple, le méthylphénidate, les amphétamines ou la cocaïne). Les patients doivent être surveillés afin de détecter tout signe de mauvaise utilisation ou d'abus (par exemple, augmentation des doses ou comportement de recherche de drogue).
Le potentiel d'abus du modafinil (200, 400 et 800 mg) a été évalué par rapport au méthylphénidate (45 et 90 mg) dans une étude en milieu hospitalier chez des personnes expérimentées avec des drogues d'abus. Les résultats de cette étude clinique ont démontré que le modafinil produisait des effets et des sentiments psychoactifs et euphoriques compatibles avec d'autres stimulants du SNC programmés (méthylphénidate).
Haut
Surdosage
Expérience humaine
Aucun surdosage n'a été rapporté dans les études cliniques de NUVIGIL. Les symptômes d'un surdosage de NUVIGIL sont probablement similaires à ceux du modafinil. Le surdosage dans les essais cliniques sur le modafinil comprenait l'excitation ou l'agitation, l'insomnie et des élévations légères ou modérées des paramètres hémodynamiques. D'après l'expérience post-commercialisation du modafinil, aucun surdosage mortel n'a été signalé avec le modafinil seul (doses allant jusqu'à 12 grammes). Les surdoses impliquant plusieurs médicaments, dont le modafinil, ont entraîné des issues fatales. Les symptômes accompagnant le plus souvent une surdose de modafinil, seuls ou en combinaison avec d'autres médicaments, ont inclus; insomnie; symptômes du système nerveux central tels que l'agitation, la désorientation, la confusion, l'excitation et l'hallucination; changements digestifs tels que nausées et diarrhée; et des changements cardiovasculaires tels que la tachycardie, la bradycardie, l'hypertension et les douleurs thoraciques.
Gestion des surdosages
Il n'existe aucun antidote spécifique pour les effets toxiques d'un surdosage de NUVIGIL. Ces surdosages doivent être gérés avec des soins principalement de soutien, y compris une surveillance cardiovasculaire. En l'absence de contre-indications, des vomissements provoqués ou un lavage gastrique doivent être envisagés. Il n'y a pas de données suggérant l'utilité de la dialyse ou de l'acidification ou de l'alcalinisation urinaire pour améliorer l'élimination des médicaments. Le médecin doit envisager de contacter un centre anti-poison pour obtenir des conseils sur le traitement de tout surdosage.
Haut
Dosage et administration
Syndrome d'apnée / hypopnée obstructive du sommeil (SAHOS) et narcolepsie
La dose recommandée de NUVIGIL pour les patients atteints de SAHOS ou de narcolepsie est de 150 mg ou 250 mg administrés en une seule dose le matin. Chez les patients atteints de SAHOS, des doses allant jusqu'à 250 mg / jour, administrées en une seule dose, ont été bien tolérées, mais Il n'y a pas de preuve cohérente que cette dose confère un avantage supplémentaire au-delà de la dose de 150 mg / jour (voir Pharmacologie clinique et Essais cliniques).
Trouble du sommeil au travail posté (SWSD)
La dose recommandée de NUVIGIL pour les patients atteints de SWSD est de 150 mg administrés quotidiennement environ 1 heure avant le début de leur quart de travail.
Un ajustement posologique doit être envisagé pour les médicaments concomitants qui sont des substrats du CYP3A4 / 5, tels que les contraceptifs stéroïdiens, le triazolam et la cyclosporine (voir PRÉCAUTIONS, Interactions médicamenteuses).
Les médicaments qui sont en grande partie éliminés par le métabolisme du CYP2C19, tels que le diazépam, le propranolol et la phénytoïne peuvent avoir élimination prolongée lors de la co-administration avec NUVIGIL et peut nécessiter une réduction de la posologie et une surveillance de la toxicité (Voir Précautions, Interactions médicamenteuses).
Chez les patients présentant une insuffisance hépatique sévère, NUVIGIL doit être administré à dose réduite (voir Pharmacologie clinique et Précautions).
Les informations sont insuffisantes pour déterminer l'innocuité et l'efficacité du dosage chez les patients présentant une insuffisance rénale sévère (voir Pharmacologie clinique et Précautions).
Chez les patients âgés, l'élimination de l'armodafinil et de ses métabolites peut être réduite en raison du vieillissement. Par conséquent, il convient d'envisager l'utilisation de doses plus faibles dans cette population (voir Pharmacologie clinique et Précautions).
Haut
Comment fournis / stockage et manutention
Nuvigil® (armodafinil) Comprimés [C-IV]
50 mg: Chaque comprimé rond, blanc à blanc cassé est gravé  d'un côté et "205" de l'autre.
d'un côté et "205" de l'autre.
NDC 63459-205-60 - Bouteilles de 60
150 mg: Chaque comprimé ovale, blanc à blanc cassé est gravé d'un côté et "215" de l'autre.
NDC 63459-215-60 - Bouteilles de 60
250 mg: Chaque comprimé ovale, blanc à blanc cassé est gravé d'un côté et "225" de l'autre.
NDC 63459-225-60 - Bouteilles de 60
Conserver à 20 ° - 25 ° C (68 ° - 77 ° F).
Fabriqué pour:
Cephalon, Inc.
Frazer, PA 19355
dernière mise à jour 02/2010
Fiche d'information patient Nuvigil (en anglais simple)
Informations détaillées sur Signes, symptômes, causes, traitements des troubles du sommeil
Les informations contenues dans cette monographie ne sont pas destinées à couvrir toutes les utilisations, directions, précautions, interactions médicamenteuses ou effets indésirables possibles. Ces informations sont généralisées et ne sont pas destinées à un avis médical spécifique. Si vous avez des questions sur les médicaments que vous prenez ou si vous souhaitez plus d'informations, consultez votre médecin, votre pharmacien ou votre infirmière.
retour à:
~ tous les articles sur les troubles du sommeil


