
Avandia pour le traitement du diabète
Nom de la marque: AVANDIA
Nom générique: maléate de rosiglitazone
Contenu:
Indications et usage
Dosage et administration
Formes posologiques et forces
Contre-indications
Avertissements et precautions
Effets indésirables
Interactions médicamenteuses
Utilisation dans des populations spécifiques
Surdosage
La description
Pharmacologie clinique
Toxicologie non clinique
Etudes cliniques
Comment fournie
Avandia, maléate de rosiglitazone, informations destinées aux patients (en anglais simple)
AVERTISSEMENT
INSUFFISANCE CARDIAQUE CONGESTIVE ET ISCHÉMIE MYOCARDIQUE
- Les thiazolidinediones, y compris la rosiglitazone, provoquent ou aggravent une insuffisance cardiaque congestive chez certains patients [voir AVERTISSEMENTS ET PRECAUTIONS]. Après l'initiation d'AVANDIA et après une augmentation de la dose, observer attentivement les patients pour détecter les signes et symptômes d'insuffisance cardiaque (y compris une prise de poids excessive et rapide, une dyspnée et / ou un œdème). Si ces signes et symptômes se développent, l'insuffisance cardiaque doit être gérée conformément aux normes de soins actuelles. De plus, l'arrêt ou la réduction de la dose d'AVANDIA doit être envisagé.
- AVANDIA n'est pas recommandé chez les patients présentant une insuffisance cardiaque symptomatique. L'initiation d'AVANDIA chez les patients présentant une insuffisance cardiaque NYHA de classe III ou IV est contre-indiquée. [Voir CONTRE-INDICATIONS et AVERTISSEMENTS ET PRECAUTIONS.]
- Une méta-analyse de 42 études cliniques (durée moyenne 6 mois; 14 237 patients au total), dont la plupart comparaient AVANDIA au placebo, ont montré qu'AVANDIA était associé à un risque accru d'événements ischémiques myocardiques tels que l'angine de poitrine ou l'infarctus du myocarde. Trois autres études (durée moyenne 41 mois; 14 067 patients au total), comparant AVANDIA à certains autres antidiabétiques oraux approuvés ou à un placebo, n'ont pas confirmé ou exclu ce risque. Dans leur intégralité, les données disponibles sur le risque d'ischémie myocardique ne sont pas concluantes. [Voir AVERTISSEMENTS ET PRECAUTIONS.]
Haut
Indications et usage
Monothérapie et thérapie combinée
AVANDIA est indiqué en complément du régime alimentaire et de l'exercice pour améliorer le contrôle glycémique chez les adultes atteints de diabète sucré de type 2.
Limitations d'utilisation importantes
- En raison de son mécanisme d'action, AVANDIA n'est actif qu'en présence d'insuline endogène. Par conséquent, AVANDIA ne doit pas être utilisé chez les patients atteints de diabète sucré de type 1 ou pour le traitement de l'acidocétose diabétique.
- La co-administration d'AVANDIA et d'insuline n'est pas recommandée.
- L'utilisation d'AVANDIA avec des nitrates n'est pas recommandée.
Haut
Dosage et administration
La gestion du traitement antidiabétique doit être individualisée. Tous les patients doivent commencer AVANDIA à la dose recommandée la plus faible. D'autres augmentations de la dose d'AVANDIA doivent être accompagnées d'une surveillance attentive des événements indésirables liés à la rétention hydrique [voir Avertissement encadré et Avertissements et precautions].
AVANDIA peut être administré à une dose initiale de 4 mg soit en une dose quotidienne unique, soit en 2 doses divisées. Pour les patients qui répondent de manière inadéquate après 8 à 12 semaines de traitement, comme déterminé par la réduction de la glycémie à jeun (FPG), la dose peut être augmentée à 8 mg par jour en monothérapie ou en association avec la metformine, la sulfonylurée ou la sulfonylurée plus metformine. Les réductions des paramètres glycémiques par la dose et le régime sont décrites sous Études cliniques. AVANDIA peut être pris avec ou sans nourriture.
La dose quotidienne totale d'AVANDIA ne doit pas dépasser 8 mg.
Monothérapie
La dose initiale habituelle d'AVANDIA est de 4 mg, administrée soit en dose unique une fois par jour, soit en doses fractionnées deux fois par jour. Dans les essais cliniques, le schéma posologique de 4 mg deux fois par jour a entraîné la plus grande réduction du FPG et de l'hémoglobine A1c (HbA1c).
Combinaison avec la sulfonylurée ou la metformine
Lorsque AVANDIA est ajouté à un traitement existant, la ou les doses actuelles d'agent (s) peuvent être poursuivies au début du traitement par AVANDIA.
Sulfonylurée: Lorsqu'elle est utilisée en association avec la sulfonylurée, la dose initiale habituelle d'AVANDIA est de 4 mg administrée soit en une dose unique une fois par jour, soit en doses fractionnées deux fois par jour. Si les patients rapportent une hypoglycémie, la dose de sulfonylurée doit être diminuée.
Metformine: La dose initiale habituelle d'AVANDIA en association avec la metformine est de 4 mg administrés soit en dose unique une fois par jour, soit en doses fractionnées deux fois par jour. Il est peu probable que la dose de metformine nécessite un ajustement en raison d'une hypoglycémie pendant le traitement d'association avec AVANDIA.
Combinaison avec Sulfonylurea Plus Metformin
La dose initiale habituelle d'AVANDIA en association avec une sulfonylurée et de la metformine est de 4 mg administrés soit en une dose unique une fois par jour, soit en doses fractionnées deux fois par jour. Si les patients rapportent une hypoglycémie, la dose de sulfonylurée doit être diminuée.
Populations de patients spécifiques
Insuffisance rénale: Aucun ajustement posologique n'est nécessaire lorsque AVANDIA est utilisé en monothérapie chez des patients atteints d'insuffisance rénale. Étant donné que la metformine est contre-indiquée chez ces patients, l'administration concomitante de metformine et d'AVANDIA est également contre-indiquée chez les patients atteints d'insuffisance rénale.
Insuffisance hépatique: Les enzymes hépatiques doivent être mesurées avant de commencer le traitement par AVANDIA. Le traitement par AVANDIA ne doit pas être instauré si le patient présente des signes cliniques de maladie du foie ou augmentation des taux de transaminases sériques (ALT> 2,5X limite supérieure de la normale au début de thérapie). Après l'initiation d'AVANDIA, les enzymes hépatiques doivent être surveillées périodiquement selon le jugement clinique du professionnel de la santé. [Voir Avertissements et precautions et PHARMACOLOGIE CLINIQUE.]
Pédiatrique: Les données sont insuffisantes pour recommander l'utilisation pédiatrique d'AVANDIA [voir UTILISATION DANS DES POPULATIONS SPÉCIFIQUES].
Haut
Formes posologiques et forces
Le comprimé pentagonal pelliculé TILTAB contient de la rosiglitazone sous forme de maléate comme suit:
- 2 mg - rose, en creux avec SB d'un côté et 2 de l'autre
- 4 mg - orange, en creux avec SB d'un côté et 4 de l'autre
- 8 mg - brun-rouge, en creux avec SB d'un côté et 8 de l'autre
Haut
Contre-indications
L'initiation d'AVANDIA chez les patients atteints d'insuffisance cardiaque de classe III ou IV établie par la New York Heart Association (NYHA) est contre-indiquée [voir AVERTISSEMENT EN BOÎTE].
Haut
Avertissements et precautions
Insuffisance cardiaque
AVANDIA, comme les autres thiazolidinediones, seul ou en association avec d'autres agents antidiabétiques, peut provoquer une rétention hydrique qui peut exacerber ou conduire à une insuffisance cardiaque. Les patients doivent être surveillés pour détecter les signes et symptômes d'insuffisance cardiaque. Si ces signes et symptômes se développent, l'insuffisance cardiaque doit être gérée conformément aux normes de soins actuelles. De plus, l'arrêt ou la réduction de la dose de rosiglitazone doit être envisagé [voir AVERTISSEMENT EN BOÎTE].
Les patients atteints d'insuffisance cardiaque congestive (CHF) NYHA classe I et II traités avec AVANDIA ont un risque accru d'événements cardiovasculaires. Une étude échocardiographique de 52 semaines, en double aveugle, contrôlée contre placebo a été menée chez 224 patients de type 2 diabète sucré et NYHA classe I ou II CHF (fraction d'éjection â ‰ ¤ 45%) sur antidiabétique de fond et CHF thérapie. Un comité indépendant a mené une évaluation en aveugle des événements liés aux fluides (y compris insuffisance cardiaque congestive) et hospitalisations cardiovasculaires selon des critères prédéfinis (arbitrage). Indépendamment de l'arbitrage, d'autres événements indésirables cardiovasculaires ont été signalés par les enquêteurs. Bien qu'aucune différence de traitement dans le changement par rapport à la ligne de base des fractions d'éjection n'ait été observée, plus des événements indésirables cardiovasculaires ont été observés après le traitement par AVANDIA par rapport au placebo pendant la Étude de 52 semaines. (Voir le tableau 1.)
Tableau 1. Événements indésirables cardiovasculaires émergents chez les patients atteints d'insuffisance cardiaque congestive (NYHA classe I et II) traités avec AVANDIA ou Placebo (en plus du traitement antidiabétique et CHF de fond)
| Événements | AVANDIA N = 110 n (%) |
Placebo N = 114 n (%) |
| Jugé | ||
| Décès cardiovasculaires | 5 (5%) | 4 (4%) |
| CHF s'aggrave | 7 (6%) | 4 (4%) |
| - avec hospitalisation de nuit | 5 (5%) | 4 (4%) |
| - sans hospitalisation de nuit | 2 (2%) | 0 (0%) |
| Oedème nouveau ou aggravé | 28 (25%) | 10 (9%) |
| Dyspnée nouvelle ou aggravée | 29 (26%) | 19 (17%) |
| Augmentation des médicaments contre l'ICC | 36 (33%) | 20 (18%) |
| Hospitalisation cardiovasculaire * | 21 (19%) | 15 (13%) |
| Déclaré par l'investigateur, non jugé | ||
| Événements indésirables ischémiques | 10 (9%) | 5 (4%) |
| - Infarctus du myocarde | 5 (5%) | 2 (2%) |
| - Angine | 6 (5%) | 3 (3%) |
| * Comprend l'hospitalisation pour toute raison cardiovasculaire. |
L'initiation d'AVANDIA chez les patients présentant une insuffisance cardiaque NYHA de classe III ou IV est contre-indiquée. AVANDIA n'est pas recommandé chez les patients présentant une insuffisance cardiaque symptomatique. [Voir AVERTISSEMENT EN BOÎTE.]
Les patients souffrant de syndromes coronariens aigus n'ont pas été étudiés dans des essais cliniques contrôlés. Compte tenu du potentiel de développement de l'insuffisance cardiaque chez les patients ayant un événement coronarien aigu, l'initiation d'AVANDIA n'est pas recommandé pour les patients souffrant d'un événement coronarien aigu, et l'arrêt d'AVANDIA pendant cette phase aiguë doit être pris en considération.
Les patients avec un statut cardiaque NYHA de classe III et IV (avec ou sans CHF) n'ont pas été étudiés dans des essais cliniques contrôlés. AVANDIA n'est pas recommandé chez les patients ayant un état cardiaque NYHA de classe III et IV.
Ischémie myocardique
Méta-analyse de l'ischémie myocardique dans un groupe de 42 essais cliniques
Une méta-analyse a été réalisée rétrospectivement pour évaluer les événements indésirables cardiovasculaires signalés dans 42 essais cliniques contrôlés, randomisés et à double insu (durée moyenne de 6 mois).1
Ces études avaient été menées pour évaluer l'efficacité hypoglycémiante dans le diabète de type 2, et aucune évaluation prospective planifiée des événements cardiovasculaires ne s'était produite dans les essais. Certains essais étaient contrôlés par placebo et certains utilisaient des antidiabétiques oraux actifs comme témoins. Les études contrôlées par placebo comprenaient des essais en monothérapie (monothérapie avec AVANDIA versus placebo en monothérapie) et des essais complémentaires (AVANDIA ou placebo, ajoutés à la sulfonylurée, la metformine ou l'insuline). Les études de contrôle actif comprenaient des essais en monothérapie (monothérapie avec AVANDIA versus sulfonylurée ou metformine en monothérapie) et des essais complémentaires (AVANDIA plus sulfonylurée ou AVANDIA plus metformine, contre sulfonylurée plus metformine). Au total, 14 237 patients ont été inclus (8 604 dans les groupes de traitement contenant AVANDIA, 5 633 groupes de comparaison), avec 4 143 années-patients d'exposition à AVANDIA et 2 675 années-patients d'exposition à AVANDIA comparateur. Les événements ischémiques myocardiques incluaient une angine de poitrine, une angine de poitrine aggravée, une angine instable, un arrêt cardiaque, des douleurs thoraciques, une coronaire occlusion artérielle, dyspnée, infarctus du myocarde, thrombose coronaire, ischémie myocardique, maladie coronarienne et artère coronaire désordre. Dans cette analyse, un risque accru d'ischémie myocardique avec AVANDIA par rapport aux comparateurs groupés a été observée (2% d'AVANDIA contre 1,5% de comparateurs, odds ratio 1,4, intervalle de confiance à 95% [IC] 1,1, 1.8). Un risque accru d'événements ischémiques myocardiques avec AVANDIA a été observé dans les études contrôlées versus placebo, mais pas dans les études contrôlées actives. (Voir figure 1.)
Un risque accru accru d'événements ischémiques myocardiques a été observé dans les études où AVANDIA a été ajouté à l'insuline (2,8% pour AVANDIA plus insuline contre 1,4% pour le placebo plus insuline, [OR 2,1, IC 95% 0,9, 5.1]). Ce risque accru reflète une différence de 3 événements pour 100 patients-années (IC à 95% -0,1, 6,3) entre les groupes de traitement. [Voir AVERTISSEMENTS ET PRECAUTIONS.]
Figure 1. Diagramme forestier des rapports de cotes (intervalles de confiance à 95%) pour les événements ischémiques myocardiques dans la méta-analyse de 42 essais cliniques

Un risque accru accru d'ischémie myocardique a également été observé chez les patients ayant reçu AVANDIA et un traitement de fond par les nitrates. Pour AVANDIA (N = 361) par rapport au témoin (N = 244) chez les utilisateurs de nitrate, le rapport de cotes était de 2,9 (IC à 95% 1,4, 5,9), tandis que pour les non-utilisateurs de nitrate (environ 14 000 patients au total), le rapport de cotes était de 1,3 (IC à 95% 0,9, 1.7). Ce risque accru représente une différence de 12 événements ischémiques myocardiques pour 100 patients-années (IC à 95% 3,3, 21,4). La plupart des utilisateurs de nitrate avaient établi une maladie coronarienne. Chez les patients atteints d'une maladie coronarienne connue qui ne suivaient pas de traitement aux nitrates, un risque accru d'événements ischémiques myocardiques pour AVANDIA par rapport au comparateur n'a pas été démontré.
Événements ischémiques myocardiques dans les grands essais contrôlés randomisés prospectifs à long terme d'AVANDIA
Les données de 3 autres grands essais cliniques à long terme, prospectifs, randomisés et contrôlés d'AVANDIA ont été évaluées séparément de la méta-analyse. Ces 3 essais comprennent un total de 14 067 patients (groupes de traitement contenant AVANDIA N = 6 311, comparateur N = 7 756), avec une exposition patient-année de 21 803 patients-années pour AVANDIA et de 25 998 patients-années pour comparateur. La durée du suivi a dépassé 3 ans dans chaque étude. ADOPT (A Diabetes Outcome Progression Trial) était une étude randomisée et contrôlée de 4 à 6 ans menée auprès de patients récemment diagnostiqués atteints de diabète de type 2 naïfs de traitement médicamenteux.
Il s'agissait d'un essai d'efficacité et de sécurité générale conçu pour examiner la durabilité de
AVANDIA en monothérapie (N = 1 456) pour le contrôle glycémique du diabète de type 2, avec bras de comparaison de sulfonylurée en monothérapie (N = 1 441) et de metformine en monothérapie (N = 1 454). DREAM (Diabetes Reduction Assessment with Rosiglitazone and Ramipril Medication, published report2) était une Étude randomisée contrôlée contre placebo de 5 ans chez des patients présentant une tolérance au glucose altérée et / ou un jeûne altéré glucose. Il avait un plan factoriel 2x2, destiné à évaluer l'effet d'AVANDIA, et séparément du ramipril (un inhibiteur de l'enzyme de conversion de l'angiotensine [ACEI]), sur la progression vers un diabète manifeste. Dans DREAM, 2 635 patients étaient dans des groupes de traitement contenant AVANDIA et 2 634 étaient dans des groupes de traitement ne contenant pas AVANDIA. AVANDIA.Des résultats intermédiaires ont été publiés 3 pour RECORD (Rosiglitazone Evaluated for Cardiac Outcome and Regulation of Glycemia in Diabetes), une étude ouverte et continue sur les résultats cardiovasculaires de 6 ans chez les patients atteints de diabète de type 2 avec un traitement moyen durée de 3,75 ans. RECORD comprend les patients qui ont échoué en monothérapie à la metformine ou aux sulfonylurées; ceux qui ont échoué à la metformine sont randomisés pour recevoir soit l'add-on AVANDIA soit l'add-on sulfonylurée, et ceux qui ont échoué sulfonylurée sont randomisés pour recevoir soit add-on AVANDIA ou add-on metformine. Dans RECORD, un total de 2 220 patients reçoivent un complément AVANDIA et 2 227 patients sont sous l'un des schémas complémentaires ne contenant pas AVANDIA.
Pour ces 3 essais, des analyses ont été réalisées à l'aide d'un composite des événements cardiovasculaires indésirables majeurs (infarctus du myocarde, décès cardiovasculaire ou accident vasculaire cérébral), ci-après dénommé MACE. Ce critère d'évaluation différait du critère d'évaluation général de la méta-analyse des événements ischémiques myocardiques, dont plus de la moitié étaient des angines. L'infarctus du myocarde incluait un infarctus du myocarde fatal et non fatal, ainsi qu'une mort subite. Comme le montre la figure 2, les résultats pour les 3 critères d'évaluation (MACE, MI et mortalité totale) n'étaient pas statistiquement significativement différents entre AVANDIA et les comparateurs.
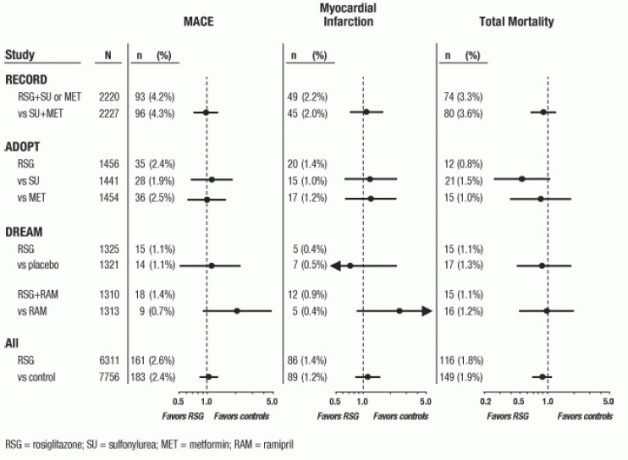
Dans les analyses préliminaires de l'essai DREAM, l'incidence des événements cardiovasculaires était plus élevée chez les sujets qui AVANDIA en association avec le ramipril que chez les sujets ayant reçu du ramipril seul, comme illustré dans Figure 2. Cette constatation n'a pas été confirmée dans ADOPT et RECORD (essais contrôlés actifs chez des patients atteints de diabète), dans lesquels 30% et 40% des patients, respectivement, ont signalé l'utilisation d'inhibiteurs de l'ECA au départ.
Dans leur intégralité, les données disponibles sur le risque d'ischémie myocardique ne sont pas concluantes. Les conclusions définitives concernant ce risque attendent l'achèvement d'une étude des résultats cardiovasculaires bien conçue.
Il n'y a eu aucune étude clinique établissant des preuves concluantes d'une réduction du risque macrovasculaire avec AVANDIA ou tout autre médicament antidiabétique oral.
Insuffisance cardiaque congestive et ischémie myocardique pendant l'administration concomitante d'AVANDIA avec de l'insuline
Dans les études dans lesquelles AVANDIA a été ajouté à l'insuline, AVANDIA a augmenté le risque d'insuffisance cardiaque congestive et d'ischémie myocardique. (Voir le tableau 2.)
L'administration concomitante d'AVANDIA et d'insuline n'est pas recommandée. [Voir Indications et usage et AVERTISSEMENTS ET PRECAUTIONS.]
Dans cinq, 26 semaines, des essais contrôlés, randomisés, en double aveugle qui ont été inclus dans la méta-analyse [voir AVERTISSEMENTS ET PRECAUTIONS], les patients atteints de diabète sucré de type 2 ont été randomisés pour une co-administration d'AVANDIA et d'insuline (N = 867) ou d'insuline (N = 663). Dans ces 5 essais, AVANDIA a été ajouté à l'insuline. Ces essais ont inclus des patients atteints de diabète de longue date (durée médiane de 12 ans) et d'une prévalence élevée de conditions médicales, y compris neuropathie périphérique, rétinopathie, cardiopathie ischémique, maladie vasculaire et cœur congestif échec. Le nombre total de patients présentant une insuffisance cardiaque congestive émergente était de 21 (2,4%) et 7 (1,1%) dans les groupes AVANDIA plus insuline et insuline, respectivement. Le nombre total de patients présentant une ischémie myocardique émergente était de 24 (2,8%) et 9 (1,4%) dans les groupes AVANDIA plus insuline et insuline, respectivement (OR 2,1 [IC 95% 0,9, 5,1]). Bien que le taux d'événements d'insuffisance cardiaque congestive et d'ischémie myocardique ait été faible dans les études dans la population, le taux d'événements était systématiquement 2 fois ou plus avec la co-administration d'AVANDIA et insuline. Ces événements cardiovasculaires ont été notés aux doses quotidiennes de 4 mg et de 8 mg d'AVANDIA. (Voir le tableau 2.)
Tableau 2. Occurrence d'événements cardiovasculaires dans 5 essais contrôlés d'addition d'AVANDIA à un traitement par insuline établi
| Un événement* | AVANDIA + Insuline (n = 867) n (%) |
Insuline (n = 663) n (%) |
| Insuffisance cardiaque congestive | 21 (2.4%) | 7 (1.1%) |
| Ischémie myocardique | 24 (2.8%) | 9 (1.4%) |
| Composite de décès cardiovasculaire, d'infarctus du myocarde ou d'accident vasculaire cérébral | 10 (1.2%) | 5 (0.8%) |
| Accident vasculaire cérébral | 5 (0.6%) | 4 (0.6%) |
| Infarctus du myocarde | 4 (0.5%) | 1 (0.2%) |
| Mort cardiovasculaire | 4 (0.5%) | 1 (0.2%) |
| Tous les décès | 6 (0.7%) | 1 (0.2%) |
| * Les événements ne sont pas exclusifs; c.-à-d., un patient avec une mort cardiovasculaire due à un infarctus du myocarde serait compté dans 4 catégories d'événements (ischémie myocardique; décès cardiovasculaire, infarctus du myocarde ou accident vasculaire cérébral; infarctus du myocarde; mort cardiovasculaire). |
Dans un sixième essai contrôlé randomisé en double aveugle de 24 semaines sur AVANDIA et l'administration concomitante d'insuline, de l'insuline a été ajoutée à AVANDAMET® (maléate de rosiglitazone et chlorhydrate de metformine) (n = 161) et par rapport à l'insuline plus le placebo (n = 158), après un rodage en simple aveugle de 8 semaines avec AVANDAMET. Les patients présentant un œdème nécessitant un traitement pharmacologique et ceux souffrant d'insuffisance cardiaque congestive ont été exclus au départ et pendant la période de rodage.
Dans le groupe recevant AVANDAMET plus insuline, il y a eu un événement ischémique myocardique et une mort subite. Aucune ischémie myocardique n'a été observée dans le groupe insuline et aucune insuffisance cardiaque congestive n'a été signalée dans les deux groupes de traitement.
Œdème
AVANDIA doit être utilisé avec prudence chez les patients présentant un œdème. Dans une étude clinique menée auprès de volontaires sains ayant reçu 8 mg d'AVANDIA une fois par jour pendant 8 semaines, il y a eu une augmentation statistiquement significative du volume plasmatique médian par rapport au placebo.
Étant donné que les thiazolidinediones, y compris la rosiglitazone, peuvent provoquer une rétention hydrique, qui peut exacerber ou conduire à une insuffisance cardiaque congestive, AVANDIA doit être utilisé avec prudence chez les patients à risque échec. Les patients doivent être surveillés pour détecter les signes et symptômes d'insuffisance cardiaque [voir AVERTISSEMENT EN BOÎTE, AVERTISSEMENTS ET PRECAUTIONS ].
Dans les essais cliniques contrôlés de patients atteints de diabète de type 2, un œdème léger à modéré a été signalé chez des patients traités par AVANDIA et peut être lié à la dose. Les patients présentant un œdème en cours étaient plus susceptibles de présenter des événements indésirables associés à un œdème s'ils étaient sous traitement d'association avec l'insuline et AVANDIA [voir EFFETS INDÉSIRABLES].
Gain de poids
Une prise de poids liée à la dose a été observée avec AVANDIA seul et en association avec d'autres hypoglycémiants (tableau 3). Le mécanisme de prise de poids n'est pas clair mais implique probablement une combinaison de rétention d'eau et d'accumulation de graisse.
Dans l'expérience post-commercialisation, des augmentations de poids inhabituellement rapides et des augmentations supérieures à celles généralement observées dans les essais cliniques ont été rapportées. Les patients qui connaissent de telles augmentations doivent être évalués pour l'accumulation de liquide et les événements liés au volume tels que l'oedème excessif et l'insuffisance cardiaque congestive [voir AVERTISSEMENT EN BOÎTE].
Tableau 3. Changements de poids (kg) par rapport à la ligne de base au point final pendant les essais cliniques
| Monothérapie | Durée | Groupe de contrôle | AVANDIA 4 mg Médian (25e, 75e centile) |
AVANDIA 8 mg Médian (25e, 75e centile) |
|
| Médian (25e, 75e centile) | |||||
| 26 semaines | placebo | -0.9 (-2.8, 0.9) n = 210 |
1.0 (-0.9, 3.6) n = 436 |
3.1 (1.1, 5.8) n = 439 |
|
| 52 semaines | sulfonylurée | 2.0 (0, 4.0) n = 173 |
2.0 (-0.6, 4.0) n = 150 |
2.6 (0, 5.3) n = 157 |
|
| Thérapie combinée | |||||
| Sulfonylurée | 24-26 semaines | sulfonylurée | 0 (-1.0, 1.3) n = 1 155 |
2.2 (0.5, 4.0) n = 613 |
3.5 (1.4, 5.9) n = 841 |
| Metformine | 26 semaines | metformine | -1.4 (-3.2, 0.2) n = 175 |
0.8 (-1.0, 2.6) n = 100 |
2.1 (0, 4.3) n = 184 |
| Insuline | 26 semaines | insuline | 0.9 (-0.5, 2.7) n = 162 |
4.1 (1.4, 6.3) n = 164 |
5.4 (3.4, 7.3) n = 150 |
| Sulfonylurée + metformine | 26 semaines | sulfonylurée + metformine | 0.2 (-1.2, 1.6) n = 272 |
2.5 (0.8, 4.6) n = 275 |
4.5 (2.4, 7.3) n = 276 |
Dans un essai comparatif en monothérapie de 4 à 6 ans (ADOPT) chez des patients récemment diagnostiqués avec un diabète de type 2 non traités auparavant avec des médicaments antidiabétiques [voir Étude cliniques], la variation du poids médian (25e, 75e centiles) par rapport à l'inclusion à 4 ans était de 3,5 kg (0,0, 8,1) pour AVANDIA, 2,0 kg (-1,0, 4,8) pour le glyburide et -2,4 kg (-5,4, 0,5) pour la metformine.
Dans une étude de 24 semaines menée chez des patients pédiatriques âgés de 10 à 17 ans traités par AVANDIA 4 à 8 mg par jour, une prise de poids médiane de 2,8 kg (25e, 75e centiles: 0,0, 5,8) a été signalé.
Effets hépatiques
Les enzymes hépatiques doivent être mesurées avant le début du traitement par AVANDIA chez tous les patients et périodiquement par la suite, selon le jugement clinique du professionnel de la santé. Le traitement par AVANDIA ne doit pas être instauré chez les patients présentant des taux d'enzymes hépatiques de base augmentés (ALT> 2,5X limite supérieure de la normale). Patients avec des enzymes hépatiques modérément élevées (niveaux d'ALAT 2.5 ¤ 2,5 fois la limite supérieure de la normale) au départ ou pendant le traitement par AVANDIA doit être évalué pour déterminer la cause de l'enzyme hépatique élévation. L'initiation ou la poursuite du traitement par AVANDIA chez les patients présentant une légère élévation des enzymes hépatiques doit se faire avec prudence. et inclure un suivi clinique étroit, y compris une surveillance des enzymes hépatiques, afin de déterminer si l'élévation des enzymes hépatiques empirer. Si, à tout moment, les taux d'ALT augmentent à> 3X la limite supérieure de la normale chez les patients traités par AVANDIA, les taux d'enzymes hépatiques doivent être revérifiés dès que possible. Si les taux d'ALAT restent> 3X la limite supérieure de la normale, le traitement par AVANDIA doit être arrêté.
Si un patient développe des symptômes suggérant un dysfonctionnement hépatique, qui peuvent inclure des nausées inexpliquées, des vomissements, des douleurs abdominales, de la fatigue, de l'anorexie et / ou des urines foncées, les enzymes hépatiques doivent être contrôlées. La décision de poursuivre ou non le traitement par AVANDIA doit être guidée par un jugement clinique en attendant les évaluations de laboratoire. Si un ictère est observé, le traitement médicamenteux doit être interrompu. [Voir EFFETS INDÉSIRABLES.]
Oedème maculaire
Un œdème maculaire a été rapporté dans l'expérience post-commercialisation chez certains patients diabétiques qui prenaient AVANDIA ou une autre thiazolidinedione. Certains patients présentaient une vision floue ou une diminution de l'acuité visuelle, mais certains patients semblent avoir été diagnostiqués lors d'un examen ophtalmologique de routine. La plupart des patients avaient un œdème périphérique au moment du diagnostic d'œdème maculaire. Certains patients ont présenté une amélioration de leur œdème maculaire après l'arrêt de leur thiazolidinedione. Les patients diabétiques devraient subir des examens ophtalmologiques réguliers, conformément aux normes de soins de l'American Diabetes Association. De plus, tout diabétique qui signale tout type de symptôme visuel doit être rapidement référé à un ophtalmologiste, quels que soient les médicaments sous-jacents du patient ou d'autres signes physiques. [Voir EFFETS INDÉSIRABLES.]
Fractures
Dans une étude comparative de 4 à 6 ans (ADOPT) du contrôle glycémique avec la monothérapie chez des patients naïfs de drogue récemment diagnostiqué avec un diabète sucré de type 2, une incidence accrue de fractures osseuses a été notée chez les patientes AVANDIA. Au cours de la période de 4 à 6 ans, l'incidence des fractures osseuses chez les femmes était de 9,3% (60/645) pour AVANDIA contre 3,5% (21/605) pour le glyburide et 5,1% (30/590) pour la metformine. Cette augmentation de l'incidence a été notée après la première année de traitement et a persisté au cours de l'étude. La majorité des fractures chez les femmes qui ont reçu AVANDIA sont survenues au bras, à la main et au pied. Ces sites de fracture sont différents de ceux habituellement associés à l'ostéoporose post-ménopausique (par exemple, hanche ou colonne vertébrale). Aucune augmentation des taux de fractures n'a été observée chez les hommes traités par AVANDIA. Le risque de fracture doit être pris en compte dans la prise en charge des patients, en particulier des femmes, traités par AVANDIA, et l’attention accordée à l’évaluation et au maintien de la santé osseuse conformément aux normes se soucier.
Effets hématologiques
Des diminutions de l'hémoglobine moyenne et de l'hématocrite sont survenues d'une manière liée à la dose chez les patients adultes traités par AVANDIA [voir EFFETS INDÉSIRABLES]. Les changements observés peuvent être liés à l'augmentation du volume plasmatique observée avec le traitement par AVANDIA.
Contrôle du diabète et de la glycémie
Les patients recevant AVANDIA en association avec d'autres hypoglycémiants peuvent présenter un risque d'hypoglycémie et une réduction de la dose de l'agent concomitant peut être nécessaire.
Des mesures périodiques de la glycémie à jeun et de l'HbA1c doivent être effectuées pour surveiller la réponse thérapeutique.
Ovulation
Le traitement par AVANDIA, comme d'autres thiazolidinediones, peut entraîner une ovulation chez certaines femmes anovulatoires préménopausées. Par conséquent, ces patientes peuvent présenter un risque accru de grossesse lors de la prise d'AVANDIA [voir Utilisation dans des populations spécifiques]. Ainsi, une contraception adéquate chez les femmes préménopausées doit être recommandée. Cet effet possible n'a pas été spécifiquement étudié dans les études cliniques; par conséquent, la fréquence de cet événement n'est pas connue.
Bien qu'un déséquilibre hormonal ait été observé dans des études précliniques [voir Toxicologie non clinique], la signification clinique de cette découverte n'est pas connue. En cas de dysfonctionnement menstruel inattendu, les avantages de la poursuite du traitement par AVANDIA doivent être revus.
Haut
Effets indésirables
Expérience d'essai clinique
Adulte
Dans les essais cliniques, environ 9 900 patients atteints de diabète de type 2 ont été traités par AVANDIA.
Essais à court terme d'AVANDIA en monothérapie et en association avec d'autres agents hypoglycémiants
L'incidence et les types d'événements indésirables signalés dans les essais cliniques à court terme d'AVANDIA en monothérapie sont présentés dans le tableau 4.
Tableau 4. Événements indésirables (¥ 5% dans tous les groupes de traitement) signalés par des patients dans des essais cliniques à court terme * en double aveugle avec AVANDIA en monothérapie
| Terme préféré | AVANDIA Monothérapie N = 2 526 % |
Placebo N = 601 % |
Metformine N = 225 % |
Sulfonyluréesâ € N = 626 % |
| Infection des voies respiratoires supérieures | 9.9 | 8.7 | 8.9 | 7.3 |
| Blessure | 7.6 | 4.3 | 7.6 | 6.1 |
| Mal de crâne | 5.9 | 5.0 | 8.9 | 5.4 |
| Mal au dos | 4.0 | 3.8 | 4.0 | 5.0 |
| Hyperglycémie | 3.9 | 5.7 | 4.4 | 8.1 |
| Fatigue | 3.6 | 5.0 | 4.0 | 1.9 |
| Sinusite | 3.2 | 4.5 | 5.3 | 3.0 |
| La diarrhée | 2.3 | 3.3 | 15.6 | 3.0 |
| Hypoglycémie | 0.6 | 0.2 | 1.3 | 5.9 |
|
* Les essais à court terme variaient de 8 semaines à 1 an. †Comprend les patients recevant du glyburide (N = 514), du gliclazide (N = 91) ou du glipizide (N = 21). |
Dans l'ensemble, les types d'effets indésirables sans égard à la causalité signalés lors de l'utilisation d'AVANDIA en association avec une sulfonylurée ou de la metformine étaient similaires à celles en monothérapie avec AVANDIA.
Les événements d'anémie et d'œdème avaient tendance à être rapportés plus fréquemment à des doses plus élevées et étaient généralement d'intensité légère à modérée et ne nécessitaient généralement pas l'arrêt du traitement par AVANDIA.
Dans les études en double aveugle, une anémie a été rapportée chez 1,9% des patients recevant AVANDIA en monothérapie contre 0,7% sous placebo, 0,6% sous sulfonylurées et 2,2% sous metformine. Les rapports d'anémie étaient plus importants chez les patients traités avec une combinaison d'AVANDIA et de metformine (7,1%) et avec une combinaison de AVANDIA et une sulfonylurée plus metformine (6,7%) par rapport à la monothérapie avec AVANDIA ou en association avec une sulfonylurée (2.3%). Des taux plus faibles d'hémoglobine / hématocrite avant traitement chez les patients inclus dans les essais cliniques de l'association metformine peuvent avoir contribué au taux de déclaration d'anémie plus élevé dans ces études [voir EFFETS INDÉSIRABLES].
Dans les essais cliniques, un œdème a été signalé chez 4,8% des patients recevant AVANDIA en monothérapie, contre 1,3% sous placebo, 1,0% sous sulfonylurées et 2,2% sous metformine. Le taux de déclaration d'œdème était plus élevé pour AVANDIA 8 mg dans les associations de sulfonylurées (12,4%) que dans les autres associations, à l'exception de l'insuline. Un œdème a été signalé chez 14,7% des patients recevant AVANDIA dans les essais de combinaison d'insuline, contre 5,4% sous insuline seule. Des cas d'apparition ou d'aggravation d'une insuffisance cardiaque congestive ont été signalés à des taux de 1% pour l'insuline seule et de 2% (4 mg) et 3% (8 mg) pour l'insuline en association avec AVANDIA [voir AVERTISSEMENT EN BOÎTE et AVERTISSEMENTS ET PRECAUTIONS].
Lors d'études de thérapie combinée contrôlée avec des sulfonylurées, des symptômes d'hypoglycémie légers à modérés, qui semblent être liés à la dose, ont été rapportés. Peu de patients ont été retirés pour hypoglycémie (<1%) et quelques épisodes d'hypoglycémie ont été considérés comme graves (<1%). L'hypoglycémie a été l'événement indésirable le plus fréquemment signalé dans les essais d'association d'insuline à dose fixe, bien que peu de patients se soient retirés pour hypoglycémie (4 sur 408 pour AVANDIA plus insuline et 1 sur 203 pour insuline seul). Les taux d'hypoglycémie, confirmés par la concentration de glucose sanguin capillaire ‰ ¤ 50 mg / dL, étaient de 6% pour l'insuline seule et de 12% (4 mg) et de 14% (8 mg) pour l'insuline en association avec AVANDIA. [Voir AVERTISSEMENTS ET PRECAUTIONS.]
Essai à long terme d'AVANDIA en monothérapie
Une étude de 4 à 6 ans (ADOPT) a comparé l'utilisation d'AVANDIA (n = 1 456), de glyburide (n = 1 441) et de metformine (n = 1 454) comme monothérapie chez les patients récemment diagnostiqués avec un diabète de type 2 qui n'avaient pas été traités antérieurement avec un antidiabétique des médicaments. Le tableau 5 présente les effets indésirables sans égard à la causalité; les taux sont exprimés pour 100 patients-années (PY) d'exposition pour tenir compte des différences d'exposition aux médicaments à l'étude dans les 3 groupes de traitement.
Dans ADOPT, des fractures ont été rapportées chez un plus grand nombre de femmes traitées par AVANDIA (9,3%, 2,7 / 100 patients-années) par rapport au glyburide (3,5%, 1,3 / 100 patients-années) ou à la metformine (5,1%, 1,5 / 100 années-patients).
La majorité des fractures chez les femmes qui ont reçu de la rosiglitazone ont été signalées dans le haut du bras, la main et le pied. [Voir AVERTISSEMENTS ET PRECAUTIONS.] L'incidence observée des fractures chez les patients masculins était similaire dans les 3 groupes de traitement.
Tableau 5. Événements indésirables en cours de traitement (5 événements / 100 patients-années [AP]) dans tous les groupes de traitement signalés dans un essai clinique de 4 à 6 ans d'AVANDIA en monothérapie (ADOPT)
| AVANDIA N = 1 456 PY = 4,954 |
Glyburide N = 1 441 PY = 4244 |
Metformine N = 1 454 PY = 4,906 |
|
| Rhinopharyngite | 6.3 | 6.9 | 6.6 |
| Mal au dos | 5.1 | 4.9 | 5.3 |
| Arthralgie | 5.0 | 4.8 | 4.2 |
| Hypertension | 4.4 | 6.0 | 6.1 |
| Infection des voies respiratoires supérieures | 4.3 | 5.0 | 4.7 |
| Hypoglycémie | 2.9 | 13.0 | 3.4 |
| La diarrhée | 2.5 | 3.2 | 6.8 |
Pédiatrique
Avandia a été évalué pour l'innocuité dans un seul essai contrôlé activement de patients pédiatriques atteints de diabète de type 2 dans lequel 99 ont été traités avec Avandia et 101 ont été traités avec la metformine. Les effets indésirables les plus courants (> 10%) sans tenir compte du lien de causalité avec Avandia ou la metformine ont été maux de tête (17% contre 14%), nausées (4% contre 11%), rhinopharyngite (3% contre 12%) et diarrhée (1% contre 13%). Dans cette étude, un cas d'acidocétose diabétique a été rapporté dans le groupe metformine. De plus, il y avait 3 patients dans le groupe rosiglitazone qui avaient une FPG de 300300 mg / dL, 2+ cétonurie et un écart anionique élevé.
Anomalies de laboratoire
Hématologique
Des diminutions de l'hémoglobine moyenne et de l'hématocrite sont survenues d'une manière liée à la dose chez les patients adultes traités avec Avandia (diminutions moyennes dans les études individuelles jusqu'à 1,0 g / dL d'hémoglobine et jusqu'à 3,3% hématocrite). Les changements se sont produits principalement au cours des 3 premiers mois suivant le début du traitement par Avandia ou suite à une augmentation de la dose d'Avandia. L'évolution dans le temps et l'ampleur des diminutions étaient similaires chez les patients traités avec une combinaison d'Avandia et d'autres hypoglycémiants ou en monothérapie avec Avandia. Les taux d'hémoglobine et d'hématocrite avant traitement étaient plus faibles chez les patients dans les études d'association à la metformine et pourraient avoir contribué au taux plus élevé de déclaration d'anémie. Dans une seule étude chez des patients pédiatriques, des diminutions de l'hémoglobine et de l'hématocrite (diminutions moyennes de 0,29 g / dL et 0,95%, respectivement) ont été signalées. De légères diminutions de l'hémoglobine et de l'hématocrite ont également été signalées chez des patients pédiatriques traités par Avandia. Le nombre de globules blancs a également légèrement diminué chez les patients adultes traités par Avandia. La diminution des paramètres hématologiques peut être liée à l'augmentation du volume plasmatique observée avec le traitement par Avandia.
Lipides
Des modifications des lipides sériques ont été observées après le traitement par Avandia chez l'adulte [voir Pharmacologie clinique ]. De petits changements dans les paramètres lipidiques sériques ont été signalés chez des enfants traités par Avandia pendant 24 semaines.
Niveaux de transaminases sériques
Dans les études cliniques de pré-approbation chez 4 598 patients traités par Avandia (3 600 patients-années d'exposition) et sur une période de 4 à Étude de 6 ans chez 1 456 patients traités par Avandia (4 954 patients-années d'exposition), il n'y avait aucune preuve de hépatotoxicité.
Dans les essais contrôlés pré-approuvés, 0,2% des patients traités par Avandia avaient des élévations de l'ALAT> 3X la limite supérieure de la normale par rapport à 0,2% sous placebo et 0,5% sous comparateurs actifs. Les élévations d'ALAT chez les patients traités par Avandia étaient réversibles. Une hyperbilirubinémie a été observée chez 0,3% des patients traités par Avandia, contre 0,9% sous placebo et 1% chez les patients sous comparateur actif. Dans les essais cliniques de pré-approbation, il n'y a eu aucun cas de réactions médicamenteuses idiosyncrasiques entraînant une insuffisance hépatique. [Voir Avertissements et precautions]
Dans l'essai ADOPT de 4 à 6 ans, les patients traités par Avandia (4 954 patients-années d'exposition), le glyburide (4 244 patients-années d'exposition) ou la metformine (4 906 patients-années d'exposition), en monothérapie, avait le même taux d'augmentation de l'ALAT à> 3 fois la limite supérieure de la normale (0,3 pour 100 patients-années exposition).
Expérience post-commercialisation
En plus des effets indésirables rapportés lors des essais cliniques, les événements décrits ci-dessous ont été identifiés lors de l'utilisation post-approbation d'Avandia. Étant donné que ces événements sont signalés volontairement par une population de taille inconnue, il n'est pas possible d'estimer de manière fiable leur fréquence ou d'établir toujours une relation de cause à effet avec l'exposition au médicament.
Chez les patients traités par thiazolidinedione, des événements indésirables graves avec ou sans issue fatale, potentiellement liés à l'expansion du volume (p. ex. insuffisance cardiaque congestive, œdème pulmonaire et épanchements pleuraux) ont été signalés [voir Avertissement encadré et Avertissements et precautions].
Il existe des rapports post-commercialisation avec Avandia d'hépatite, une élévation des enzymes hépatiques à 3 fois ou plus limite supérieure de l'insuffisance hépatique normale et avec ou sans issue fatale, bien que la causalité n'ait pas été établi.
Une éruption cutanée, un prurit, une urticaire, un œdème de Quincke, une réaction anaphylactique et un syndrome de Stevens-Johnson ont été rarement rapportés.
Des cas d'apparition ou d'aggravation d'un œdème maculaire diabétique avec diminution de l'acuité visuelle ont également été signalés [voir Avertissements et precautions].
Haut
Interactions médicamenteuses
Inhibiteurs et inducteurs du CYP2C8
Un inhibiteur du CYP2C8 (par exemple, le gemfibrozil) peut augmenter l'ASC de la rosiglitazone et un inducteur du CYP2C8 (par exemple, la rifampicine) peut diminuer l'ASC de la rosiglitazone. Par conséquent, si un inhibiteur ou un inducteur du CYP2C8 est démarré ou arrêté pendant le traitement par la rosiglitazone, des modifications du traitement du diabète peuvent être nécessaires en fonction de la réponse clinique. [Voir PHARMACOLOGIE CLINIQUE.]
Haut
Utilisation dans des populations spécifiques
Grossesse
Catégorie de grossesse C.
Toutes les grossesses présentent un risque de fond de malformations congénitales, de perte ou d'autres effets indésirables, quelle que soit l'exposition au médicament. Ce risque de fond est accru dans les grossesses compliquées d'hyperglycémie et peut être diminué avec un bon contrôle métabolique. Il est essentiel pour les patientes atteintes de diabète ou ayant des antécédents de diabète gestationnel de maintenir un bon contrôle métabolique avant la conception et tout au long de la grossesse. Une surveillance attentive du contrôle de la glycémie est essentielle chez ces patients. La plupart des experts recommandent d'utiliser l'insuline en monothérapie pendant la grossesse pour maintenir la glycémie aussi près que possible de la normale.
Données humaines: Il a été rapporté que la rosiglitazone traverse le placenta humain et peut être détectée dans le tissu fœtal. La signification clinique de ces résultats est inconnue. Il n'y a pas d'études adéquates et bien contrôlées chez les femmes enceintes. AVANDIA ne doit pas être utilisé pendant la grossesse.
Études animales: Il n'y a eu aucun effet sur l'implantation ou l'embryon avec un traitement à la rosiglitazone au début de la gestation chez le rat, mais le traitement au cours de la gestation mi-tardive a été associé à la mort fœtale et à un retard de croissance chez les rats et les lapins. Aucune tératogénicité n'a été observée à des doses allant jusqu'à 3 mg / kg chez le rat et 100 mg / kg chez le lapin (environ 20 et 75 fois l'ASC humaine à la dose quotidienne maximale recommandée chez l'homme, respectivement). La rosiglitazone a provoqué une pathologie placentaire chez le rat (3 mg / kg / jour). Le traitement des rats pendant la gestation pendant la lactation a réduit la taille de la portée, la viabilité néonatale et la croissance postnatale, avec un retard de croissance réversible après la puberté. Pour les effets sur le placenta, l'embryon / le fœtus et la progéniture, la dose sans effet était de 0,2 mg / kg / jour chez le rat et de 15 mg / kg / jour chez le lapin. Ces niveaux sans effet représentent environ 4 fois l'ASC humaine à la dose quotidienne maximale recommandée chez l'homme. La rosiglitazone a réduit le nombre d'implantations utérines et de descendants vivants lorsque des rats femelles juvéniles ont été traitées à 40 ans. mg / kg / jour à partir de 27 jours jusqu'à la maturité sexuelle (environ 68 fois l'ASC humaine au maximum recommandé quotidiennement) dose). Le niveau sans effet était de 2 mg / kg / jour (environ 4 fois l'ASC humaine à la dose quotidienne maximale recommandée). Il n'y a eu aucun effet sur la survie ou la croissance prénatale ou postnatale.
Travail et accouchement
L'effet de la rosiglitazone sur le travail et l'accouchement chez l'homme n'est pas connu.
Mères allaitantes
Des substances liées au médicament ont été détectées dans le lait de rats en lactation. On ne sait pas si AVANDIA est excrété dans le lait maternel. Étant donné que de nombreux médicaments sont excrétés dans le lait maternel, AVANDIA ne doit pas être administré à une femme qui allaite.
Utilisation pédiatrique
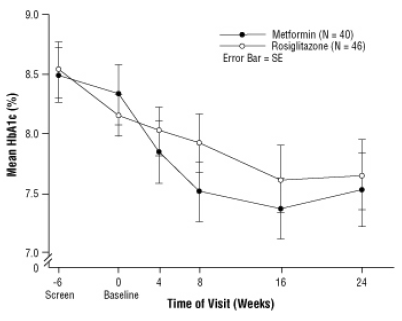
Après le placebo, y compris les conseils diététiques, les enfants atteints de diabète sucré de type 2, âgés de 10 à 17 ans et ayant un indice de masse corporelle (IMC) de base de 33 kg / m, ont été randomisés pour un traitement avec 2 mg deux fois par jour d'AVANDIA (n = 99) ou 500 mg deux fois par jour de metformine (n = 101) dans une étude clinique en double aveugle de 24 semaines procès. Comme prévu, la FPG a diminué chez les patients naïfs de médicaments contre le diabète (n = 104) et a augmenté chez les patients retirés des médicaments antérieurs (généralement la metformine) (n = 90) au cours de la période de rodage. Après au moins 8 semaines de traitement, 49% des patients traités par AVANDIA et 55% des patients traités par metformine ont vu leur dose doubler si FPG> 126 mg / dL. Pour la population globale en intention de traiter, à la semaine 24, la variation moyenne par rapport à la valeur initiale de l'HbA1c était de -0,14% avec AVANDIA et de -0,49% avec la metformine. Le nombre de patients dans cette étude était insuffisant pour établir statistiquement si ces
les effets moyens du traitement observés étaient similaires ou différents. Les effets du traitement différaient pour les patients naïfs de thérapie avec des médicaments antidiabétiques et pour les patients précédemment traités par thérapie antidiabétique (tableau 6).
Tableau 6. Changement de FPG et d'HbA1c à la semaine 24 par rapport à la dernière observation de base reportée chez les enfants ayant une HbA1c> 6,5%
| Patients naïfs | Patients déjà traités | |||
|
Metformine N = 40 |
Rosiglitazone N = 45 |
Metformine N = 43 |
Rosiglitazone N = 32 |
|
| FPG (mg / dL) | ||||
| Référence (moyenne) | 170 | 165 | 221 | 205 |
| Changement par rapport à la référence (moyenne) | -21 | -11 | -33 | -5 |
| Différence de traitement ajustée * (rosiglitazone-metformine) - (IC 95%) | 8 (-15, 30) |
21 (-9, 51) |
||
| % de patients avec une diminution de ≤ 30 mg / dL par rapport à l'inclusion | 43% | 27% | 44% | 28% |
| HbA1c (%) | ||||
| Référence (moyenne) | 8.3 | 8.2 | 8.8 | 8.5 |
| Changement par rapport à la référence (moyenne) | -0.7 | -0.5 | -0.4 | 0.1 |
| Différence de traitement ajustée * (rosiglitazone-metformine) - (IC 95%) | 0.2 (-0.6, 0.9) |
0.5 (-0.2, 1.3) |
||
| % de patients avec une baisse de 0,7% de 0,7% par rapport à l'inclusion | 63% | 52% | 54% | 31% |
|
* Les changements par rapport aux moyennes de référence sont les moindres carrés, ce qui signifie un ajustement pour l'HbA1c de base, le sexe et la région. †Des valeurs positives pour la différence favorisent la metformine. |
Les différences de traitement dépendaient de l'IMC de base ou du poids, de sorte que les effets d'AVANDIA et de la metformine semblaient plus étroitement comparables chez les patients plus lourds. Le gain de poids médian était de 2,8 kg avec la rosiglitazone et de 0,2 kg avec la metformine [voir AVERTISSEMENTS ET PRECAUTIONS]. Cinquante-quatre pour cent des patients traités par rosiglitazone et 32% des patients traités par metformine ont gagné 2 ¥ ¥ 2 kg, et 33% des patients traités par rosiglitazone et 7% des patients traités par metformine ont gagné 5 kg sur étude.
Les événements indésirables observés dans cette étude sont décrits dans Effets indésirables).
Figure 3. HbA1c moyenne au fil du temps dans une étude de 24 semaines sur AVANDIA et la metformine chez les patients pédiatriques - Sous-groupe des médicaments naïfs
Utilisation gériatrique
Les résultats de l'analyse pharmacocinétique de population ont montré que l'âge n'affecte pas significativement la pharmacocinétique de la rosiglitazone [voir PHARMACOLOGIE CLINIQUE]. Par conséquent, aucun ajustement posologique n'est nécessaire pour les personnes âgées. Dans les essais cliniques contrôlés, aucune différence globale de sécurité et d'efficacité n'a été observée entre les patients plus âgés (65 ans) et les patients plus jeunes (<65 ans).
Haut
Surdosage
Des données limitées sont disponibles concernant le surdosage chez l'homme. Dans les études cliniques menées chez des volontaires, AVANDIA a été administré à des doses orales uniques allant jusqu'à 20 mg et a été bien toléré. En cas de surdosage, un traitement de soutien approprié doit être instauré en fonction de l'état clinique du patient.
Haut
La description
AVANDIA (maléate de rosiglitazone) est un antidiabétique oral qui agit principalement en augmentant la sensibilité à l'insuline. AVANDIA améliore le contrôle glycémique tout en réduisant les niveaux d'insuline circulante.
Le maléate de rosiglitazone n'est pas lié chimiquement ou fonctionnellement aux sulfonylurées, aux biguanides ou aux inhibiteurs de l'alpha-glucosidase.
Chimiquement, le maléate de rosiglitazone est le (±) -5 - [[4- [2- (méthyl-2-pyridinylamino) éthoxy] phényl] méthyl] -2,4-thiazolidinedione, (Z) -2-butènedioate (1: 1) avec un poids moléculaire de 473,52 (357,44 base libre). La molécule a un seul centre chiral et est présente sous forme de racémate. En raison d'une interconversion rapide, les énantiomères ne peuvent pas être distingués fonctionnellement. La formule structurelle du maléate de rosiglitazone est:
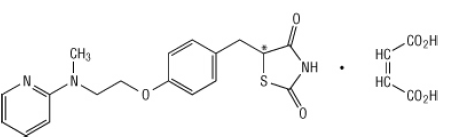
La formule moléculaire est C18H19N3O3S-C4H4O4. Le maléate de rosiglitazone est un solide blanc à blanc cassé dont le point de fusion se situe entre 122 et 123 ° C. Les valeurs de pKa du maléate de rosiglitazone sont de 6,8 et 6,1. Il est facilement soluble dans l'éthanol et un solution aqueuse tamponnée avec un pH de 2,3; la solubilité diminue avec l'augmentation du pH dans la intervalle.
Chaque comprimé pentagonal pelliculé TILTAB contient du maléate de rosiglitazone équivalent à la rosiglitazone, 2 mg, 4 mg ou 8 mg, pour administration orale. Les ingrédients inactifs sont: Hypromellose 2910, lactose monohydraté, stéarate de magnésium, cellulose microcristalline, polyéthylène glycol 3000, glycolate d'amidon sodique, dioxyde de titane, triacétine et 1 ou plusieurs des substances suivantes: oxydes de fer synthétiques rouge et jaune et talc.
Haut
Pharmacologie clinique
Mécanisme d'action
La rosiglitazone, un membre de la classe des agents antidiabétiques thiazolidinedione, améliore le contrôle glycémique en améliorant la sensibilité à l'insuline. La rosiglitazone est un agoniste hautement sélectif et puissant du récepteur gamma activé par le proliférateur des peroxysomes (PPARγ). Chez l'homme, les récepteurs PPAR se trouvent dans les tissus cibles clés pour l'action de l'insuline tels que le tissu adipeux, le muscle squelettique et le foie. L'activation des récepteurs nucléaires PPARγ régule la transcription des gènes sensibles à l'insuline impliqués dans le contrôle de la production, du transport et de l'utilisation du glucose. De plus, les gènes sensibles à PPARγ participent également à la régulation du métabolisme des acides gras.
La résistance à l'insuline est une caractéristique commune caractérisant la pathogenèse du diabète de type 2. L'activité antidiabétique de la rosiglitazone a été démontrée sur des modèles animaux de diabète de type 2 chez hyperglycémie et / ou altération de la tolérance au glucose est une conséquence de la résistance à l'insuline chez la cible tissus. La rosiglitazone réduit les concentrations de glucose dans le sang et réduit l'hyperinsulinémie chez la souris obèse ob / ob, la souris diabétique db / db et le rat Zucker gras fa / fa.
Dans les modèles animaux, l'activité antidiabétique de la rosiglitazone s'est révélée être médiée par une sensibilité accrue à l'action de l'insuline dans le foie, les muscles et les tissus adipeux. Des études pharmacologiques sur des modèles animaux indiquent que la rosiglitazone inhibe la gluconéogenèse hépatique. L'expression du transporteur de glucose régulé par l'insuline GLUT-4 a été augmentée dans le tissu adipeux. La rosiglitazone n'a pas induit d'hypoglycémie dans les modèles animaux de diabète de type 2 et / ou de tolérance au glucose altérée.
Pharmacodynamique
Les patients présentant des anomalies lipidiques n'ont pas été exclus des essais cliniques d'AVANDIA.
Dans tous les essais contrôlés de 26 semaines, dans la plage de doses recommandée, AVANDIA en monothérapie a été associé à une augmentation du cholestérol total, des LDL et des HDL et à une diminution des acides gras libres. Ces changements étaient statistiquement significativement différents des témoins placebo ou glyburide (tableau 7).
Les augmentations du LDL sont survenues principalement au cours des 1 à 2 premiers mois de traitement par AVANDIA et les niveaux de LDL sont restés supérieurs au niveau de référence tout au long des essais. En revanche, le HDL a continué d'augmenter au fil du temps. En conséquence, le rapport LDL / HDL a culminé après 2 mois de traitement et a ensuite semblé diminuer avec le temps. En raison de la nature temporelle des changements lipidiques, l'étude contrôlée par le glyburide de 52 semaines est la plus pertinente pour évaluer les effets à long terme sur les lipides. Au départ, à la semaine 26 et à la semaine 52, les rapports LDL / HDL moyens étaient respectivement de 3,1, 3,2 et 3,0 pour AVANDIA 4 mg deux fois par jour. Les valeurs correspondantes pour le glyburide étaient de 3,2, 3,1 et 2,9. Les différences de changement par rapport aux valeurs initiales entre AVANDIA et le glyburide à la semaine 52 étaient statistiquement significatives.
Le schéma des modifications des LDL et des HDL après un traitement par AVANDIA en association avec d'autres hypoglycémiants était généralement similaire à celui observé avec AVANDIA en monothérapie.
Les changements dans les triglycérides pendant le traitement par AVANDIA étaient variables et n'étaient généralement pas statistiquement différents des témoins placebo ou glyburide.
Tableau 7. Résumé des changements lipidiques moyens dans les études de 26 semaines contrôlées par placebo et de 52 semaines contrôlées par le glyburide
| Études contrôlées par placebo Semaine 26 | Études contrôlées au glyburide Semaine 26 et Semaine 52 | ||||||
| Placebo | AVANDIA | Titrage au glyburide | AVANDIA 8 mg | ||||
| 4 mg par jour * | 8 mg par jour * | Sem 26 | Sem 52 | Sem 26 | Sem 52 | ||
| Acides gras libres | |||||||
| N | 207 | 428 | 436 | 181 | 168 | 166 | 145 |
| Référence (moyenne)% | 18.1 | 17.5 | 17.9 | 26.4 | 26.4 | 26.9 | 26.6 |
| Changement par rapport à la référence (moyenne) | +0.2% | -7.8% | -14.7% | -2.4% | -4.7% | -20.8% | -21.5% |
| LDL | |||||||
| N | 190 | 400 | 374 | 175 | 160 | 161 | 133 |
| Référence (moyenne)% | 123.7 | 126.8 | 125.3 | 142.7 | 141.9 | 142.1 | 142.1 |
| Changement par rapport à la référence (moyenne) | +4.8% | +14.1% | +18.6% | -0.9% | -0.5% | +11.9% | +12.1% |
| HDL | |||||||
| N | 208 | 429 | 436 | 184 | 170 | 170 | 145 |
| Référence (moyenne)% | 44.1 | 44.4 | 43.0 | 47.2 | 47.7 | 48.4 | 48.3 |
| Changement par rapport à la référence (moyenne) | +8.0% | +11.4% | +14.2% | +4.3% | +8.7% | +14.0% | +18.5% |
| * Des groupes d'administration une fois par jour et deux fois par jour ont été combinés. |
Pharmacocinétique
La concentration plasmatique maximale (Cmax) et l'aire sous la courbe (ASC) de la rosiglitazone augmentent de manière proportionnelle à la dose sur la plage de doses thérapeutiques (tableau 8). La demi-vie d'élimination est de 3 à 4 heures et est indépendante de la dose.
Tableau 8. Paramètres pharmacocinétiques moyens (ET) de la rosiglitazone après des doses orales uniques (N = 32)
| Paramètre | 1 mg à jeun | 2 mg à jeun | 8 mg à jeun | 8 mg nourris |
| AUC0-inf [ng-h / ml] |
358 (112) |
733 (184) |
2,971 (730) |
2,890 (795) |
| Cmax [ng / ml] |
76 (13) |
156 (42) |
598 (117) |
432 (92) |
| Demi vie [heure] |
3.16 (0.72) |
3.15 (0.39) |
3.37 (0.63) |
3.59 (0.70) |
| CL / F * [L / h] |
3.03 (0.87) |
2.89 (0.71) |
2.85 (0.69) |
2.97 (0.81) |
| * CL / F = clairance orale. |
Absorption
La biodisponibilité absolue de la rosiglitazone est de 99%. Les concentrations plasmatiques maximales sont observées environ 1 heure après l'administration. L'administration de rosiglitazone avec de la nourriture n'a entraîné aucun changement dans l'exposition globale (ASC), mais il y a eu une diminution d'environ 28% de la Cmax et un retard de la Tmax (1,75 heure). Ces changements ne sont pas susceptibles d'être cliniquement significatifs; AVANDIA peut donc être administré avec ou sans nourriture.
Distribution
Le volume moyen (CV%) de distribution orale (Vss / F) de rosiglitazone est d'environ 17,6 (30%) litres, selon une analyse pharmacocinétique de population. La rosiglitazone est liée à environ 99,8% aux protéines plasmatiques, principalement à l'albumine.
Métabolisme
La rosiglitazone est largement métabolisée et aucun médicament inchangé n'est excrété dans l'urine. Les principales voies de métabolisme étaient la N-déméthylation et l'hydroxylation, suivies de la conjugaison avec du sulfate et de l'acide glucuronique. Tous les métabolites circulants sont considérablement moins puissants que les parents et, par conséquent, ne devraient pas contribuer à l'activité de sensibilisation à l'insuline de la rosiglitazone.
Les données in vitro montrent que la rosiglitazone est principalement métabolisée par l'isoenzyme 2C8 du cytochrome P450 (CYP), le CYP2C9 contribuant comme voie secondaire.
Excrétion
Après administration orale ou intraveineuse de maléate de rosiglitazone [14C], environ 64% et 23% de la dose ont été éliminés respectivement dans l'urine et les fèces. La demi-vie plasmatique du matériel apparenté au [14C] variait de 103 à 158 heures.
Pharmacocinétique de population chez les patients atteints de diabète de type 2
Analyses pharmacocinétiques de population issues de 3 grands essais cliniques incluant 642 hommes et 405 femmes diabétiques de type 2 (35 ans à 80 ans) a montré que la pharmacocinétique de la rosiglitazone n'est pas influencée par l'âge, la race, le tabagisme ou l'alcool consommation. Il a été démontré que la clairance orale (CL / F) et le volume de distribution par voie orale à l'état d'équilibre (Vss / F) augmentent avec l'augmentation du poids corporel. Sur la plage de poids observée dans ces analyses (50 à 150 kg), la plage des valeurs CL / F et Vss / F prévues a varié de <1,7 fois et <2,3 fois, respectivement.
De plus, la rosiglitazone CL / F s'est révélée être influencée à la fois par le poids et le sexe, étant plus faible (environ 15%) chez les patientes.
Populations spéciales
Gériatrique
Résultats de l'analyse pharmacocinétique de population (n = 716 <65 ans; n = 331 â 65 ans) a montré que l'âge n'affecte pas significativement la pharmacocinétique de la rosiglitazone.
Le sexe
Les résultats de l'analyse pharmacocinétique de population ont montré que la clairance orale moyenne de la rosiglitazone les patientes (n = 405) étaient environ 6% plus faibles que les patientes de même poids corporel (n = 642).
En monothérapie et en association avec la metformine, AVANDIA a amélioré le contrôle glycémique chez les hommes et les femmes. Dans les études de combinaison de metformine, l'efficacité a été démontrée sans différence de sexe dans la réponse glycémique.
Dans les études en monothérapie, une plus grande réponse thérapeutique a été observée chez les femmes; cependant, chez les patients plus obèses, les différences entre les sexes étaient moins évidentes. Pour un indice de masse corporelle (IMC) donné, les femmes ont tendance à avoir une masse grasse plus élevée que les hommes. Étant donné que la cible moléculaire PPARγ est exprimée dans les tissus adipeux, cette caractéristique de différenciation peut expliquer, au moins en partie, la plus grande réponse à AVANDIA chez les femmes. Étant donné que le traitement doit être individualisé, aucun ajustement posologique n'est nécessaire en fonction du sexe uniquement.
Insuffisance hépatique
La clairance orale non liée de la rosiglitazone était significativement plus faible chez les patients atteints d'une maladie hépatique modérée à sévère (Child-Pugh classe B / C) que chez les sujets sains. En conséquence, la Cmax non liée et l'ASC0-inf ont été augmentées respectivement de 2 et 3 fois. La demi-vie d'élimination de la rosiglitazone était d'environ 2 heures de plus chez les patients atteints d'une maladie du foie, par rapport aux sujets sains.
Le traitement par AVANDIA ne doit pas être instauré si le patient présente des signes cliniques de maladie hépatique ou augmentation des taux de transaminases sériques (ALT> 2,5 fois la limite supérieure de la normale) au départ [voir AVERTISSEMENTS ET PRECAUTIONS].
Pédiatrique
Les paramètres pharmacocinétiques de la rosiglitazone chez les patients pédiatriques ont été établis à l'aide d'une analyse pharmacocinétique de population avec des données éparses de 96 patients pédiatriques dans un seul essai clinique pédiatrique comprenant 33 hommes et 63 femmes âgés de 10 à 17 ans (poids allant de 35 à 178,3 kg). La CL / F et la V / F moyennes de la population de rosiglitazone étaient respectivement de 3,15 L / h et de 13,5 L. Ces estimations de CL / F et V / F étaient conformes aux estimations de paramètres typiques d'une analyse antérieure de la population adulte.
Insuffisance rénale
Il n'y a pas de différences cliniquement pertinentes dans la pharmacocinétique de la rosiglitazone chez les patients à une insuffisance rénale sévère ou chez les patients hémodialysés dépendants par rapport aux sujets ayant une une fonction. Aucun ajustement posologique n'est donc nécessaire chez ces patients recevant AVANDIA. Étant donné que la metformine est contre-indiquée chez les patients atteints d'insuffisance rénale, la co-administration de metformine avec AVANDIA est contre-indiquée chez ces patients.
Course
Les résultats d'une analyse pharmacocinétique de population incluant des sujets d'origine caucasienne, noire et d'autres origines ethniques indiquent que la race n'a aucune influence sur la pharmacocinétique de la rosiglitazone.
Interactions médicament-médicament
Médicaments qui inhibent, induisent ou sont métabolisés par le cytochrome P450
Des études in vitro sur le métabolisme des médicaments suggèrent que la rosiglitazone n'inhibe aucune des principales enzymes P450 à des concentrations cliniquement pertinentes. Les données in vitro montrent que la rosiglitazone est principalement métabolisée par le CYP2C8 et, dans une moindre mesure, le 2C9. AVANDIA (4 mg deux fois par jour) n'a montré aucun effet cliniquement significatif sur la pharmacocinétique de nifédipine et contraceptifs oraux (éthinylestradiol et noréthindrone), qui sont principalement métabolisés par CYP3A4.
Gemfibrozil
Administration concomitante de gemfibrozil (600 mg deux fois par jour), un inhibiteur du CYP2C8 et de rosiglitazone (4 mg une fois par jour) pendant 7 jours a augmenté l'ASC de la rosiglitazone de 127%, par rapport à l'administration de rosiglitazone (4 mg une fois par jour) seul. Étant donné le potentiel d'événements indésirables liés à la dose de rosiglitazone, une diminution de la dose de rosiglitazone peut être nécessaire lors de l'introduction du gemfibrozil [voir INTERACTIONS MÉDICAMENTEUSES].
Rifampin
L'administration de rifampicine (600 mg une fois par jour), un inducteur du CYP2C8, pendant 6 jours, diminuerait l'ASC de la rosiglitazone de 66%, par rapport à l'administration de rosiglitazone (8 mg) seule [voir INTERACTIONS MÉDICAMENTEUSES].4
Glyburide
AVANDIA (2 mg deux fois par jour) pris en concomitance avec du glyburide (3,75 à 10 mg / jour) pendant 7 jours n'a pas changé les concentrations plasmatiques moyennes de glucose à 24 heures à l'état d'équilibre chez les patients diabétiques stabilisées sous glyburide thérapie. Des doses répétées d'AVANDIA (8 mg une fois par jour) pendant 8 jours chez des sujets adultes de race blanche en bonne santé ont provoqué une diminution de l'ASC et de la Cmax du glyburide d'environ 30%. Chez les sujets japonais, l'ASC et la Cmax du glyburide ont légèrement augmenté après la co-administration d'AVANDIA.
Glimépiride
Des doses orales uniques de glimépiride chez 14 sujets adultes en bonne santé n'ont eu aucun effet cliniquement significatif sur la pharmacocinétique à l'état d'équilibre d'AVANDIA. Aucune réduction cliniquement significative de l'ASC et de la C du glimépiridemax ont été observés après des doses répétées d'AVANDIA (8 mg une fois par jour) pendant 8 jours chez des sujets adultes en bonne santé.
Metformine
L'administration concomitante d'AVANDIA (2 mg deux fois par jour) et de metformine (500 mg deux fois par jour) chez des volontaires pendant 4 jours n'ont eu aucun effet sur la pharmacocinétique à l'état d'équilibre de la metformine ou rosiglitazone.
Acarbose
L'administration concomitante d'acarbose (100 mg trois fois par jour) pendant 7 jours chez des volontaires sains n'a eu aucun effet cliniquement significatif sur la pharmacocinétique d'une dose orale unique d'AVANDIA.
Digoxine
L'administration orale répétée d'AVANDIA (8 mg une fois par jour) pendant 14 jours n'a pas modifié la pharmacocinétique à l'état d'équilibre de la digoxine (0,375 mg une fois par jour) chez des volontaires sains.
Warfarine
L'administration répétée d'AVANDIA n'a eu aucun effet cliniquement significatif sur la pharmacocinétique à l'état d'équilibre des énantiomères de la warfarine.
L'éthanol
Une administration unique d'une quantité modérée d'alcool n'a pas augmenté le risque d'hypoglycémie aiguë chez les patients atteints de diabète sucré de type 2 traités par AVANDIA.
Ranitidine
Le prétraitement à la ranitidine (150 mg deux fois par jour pendant 4 jours) n'a pas modifié la pharmacocinétique des doses orales ou intraveineuses uniques de rosiglitazone chez des volontaires sains.
Ces résultats suggèrent que l'absorption de la rosiglitazone par voie orale n'est pas modifiée dans des conditions accompagnées d'une augmentation du pH gastro-intestinal.
Haut
Toxicologie non clinique
Carcinogenèse, mutagenèse, altération de la fertilité
Cancérogenèse:
Une étude de cancérogénicité de 2 ans a été menée chez des souris Charles River CD-1 à des doses de 0,4, 1,5 et 6 mg / kg / jour chez le régime alimentaire (dose la plus élevée équivalente à environ 12 fois l'ASC humaine à la dose quotidienne maximale recommandée chez l'homme) dose). Des rats Sprague-Dawley ont été dosés pendant 2 ans par gavage oral à des doses de 0,05, 0,3 et 2 mg / kg / jour (l'équivalent de dose le plus élevé à environ 10 et 20 fois l'ASC humaine à la dose quotidienne maximale recommandée pour les rats mâles et femelles, respectivement).
La rosiglitazone n'était pas cancérogène chez la souris. Il y a eu une augmentation de l'incidence de l'hyperplasie adipeuse chez la souris à des doses de 1,5 mg / kg / jour (environ 2 fois l'ASC humaine à la dose quotidienne maximale recommandée chez l'homme). Chez le rat, il y a eu une augmentation significative de l'incidence des tumeurs bénignes du tissu adipeux (lipomes) à des doses de 0,3 mg / kg / jour (environ 2 fois l'ASC humaine à la dose quotidienne humaine maximale recommandée). Ces changements prolifératifs dans les deux espèces sont considérés en raison de la surstimulation pharmacologique persistante du tissu adipeux.
Mutagenèse:
La rosiglitazone ne s'est pas révélée mutagène ni clastogène dans les tests bactériens in vitro de mutation génique, test d'aberration chromosomique dans les lymphocytes humains, le test du micronoyau de souris in vivo et l'UDS de rat in vivo / in vitro essai. Il y avait une petite augmentation (environ 2 fois) de la mutation dans le test in vitro du lymphome de souris en présence d'une activation métabolique.
Altération de la fertilité:
La rosiglitazone n'a eu aucun effet sur l'accouplement ou la fertilité des rats mâles ayant reçu jusqu'à 40 mg / kg / jour (environ 116 fois l'ASC humaine à la dose quotidienne maximale recommandée chez l'homme). La rosiglitazone a altéré la cyclicité œstrale (2 mg / kg / jour) et réduit la fertilité (40 mg / kg / jour) des rats femelles en association avec une les taux plasmatiques de progestérone et d'estradiol (environ 20 et 200 fois l'ASC humaine à la dose quotidienne maximale recommandée chez l'homme, respectivement). Aucun effet de ce type n'a été noté à 0,2 mg / kg / jour (environ 3 fois l'ASC humaine à la dose quotidienne maximale recommandée chez l'homme). Chez les rats juvéniles traités à partir de 27 jours jusqu'à la maturité sexuelle (jusqu'à 40 mg / kg / jour), il n'y a eu aucun effet sur les performances de reproduction des mâles, ou sur la cyclicité œstrale, les performances d'accouplement ou l'incidence de la grossesse chez les femelles (environ 68 fois l'ASC humaine à la dose quotidienne maximale recommandée chez l'homme) dose). Chez les singes, la rosiglitazone (0,6 et 4,6 mg / kg / jour; environ 3 et 15 fois l'ASC humaine à la dose quotidienne humaine maximale recommandée, respectivement) a diminué l'élévation de la phase folliculaire dans l'estradiol sérique avec une réduction conséquente de la poussée hormonale lutéinisante, des niveaux plus bas de progestérone en phase lutéale, et aménorrhée. Le mécanisme de ces effets semble être une inhibition directe de la stéroïdogenèse ovarienne.
Toxicologie animale
Le poids cardiaque a augmenté chez la souris (3 mg / kg / jour), le rat (5 mg / kg / jour) et le chien (2 mg / kg / jour) avec la rosiglitazone traitements (environ 5, 22 et 2 fois l'ASC humaine à la dose quotidienne maximale recommandée chez l'homme, respectivement). Les effets chez les rats juvéniles étaient conformes à ceux observés chez les adultes. La mesure morphométrique a indiqué qu'il y avait une hypertrophie dans les tissus ventriculaires cardiaques, qui peut être due à une augmentation du travail cardiaque en raison de l'expansion du volume plasmatique.
Haut
Etudes cliniques
Monothérapie
Dans les études cliniques, le traitement par AVANDIA a entraîné une amélioration du contrôle glycémique, mesurée par le FPG et l'HbA1c, avec une réduction concomitante de l'insuline et du peptide C. Le glucose et l'insuline postprandiaux ont également été réduits. Ceci est cohérent avec le mécanisme d'action d'AVANDIA en tant que sensibilisateur à l'insuline.
La dose quotidienne maximale recommandée est de 8 mg. Des études de dosage ont suggéré qu'aucun avantage supplémentaire n'a été obtenu avec une dose quotidienne totale de 12 mg.
Études cliniques à court terme: Au total, 2 315 patients atteints de diabète de type 2, précédemment traités par un régime seul ou des médicaments antidiabétiques, ont été traités par AVANDIA en monothérapie dans 6 cas à double insu. études, qui comprenaient deux études contrôlées par placebo de 26 semaines, une étude contrôlée par glyburide de 52 semaines et 3 études de doses contrôlées par placebo d'une durée de 8 à 12 semaines. Les antidiabétiques antérieurs ont été retirés et les patients ont entamé une période de pré-placebo de 2 à 4 semaines avant la randomisation.
Deux essais de 26 semaines, en double aveugle, contrôlés contre placebo, chez des patients atteints de diabète de type 2 (n = 1 401) avec une glycémie insuffisante (FPG moyen à l'inclusion environ 228 mg / dL [101 à 425 mg / dL] et HbA1c moyenne à l'injection 8,9% [5,2% à 16,2%]), étaient menée. Le traitement par AVANDIA a produit des améliorations statistiquement significatives de la FPG et de l'HbA1c par rapport à l'inclusion et par rapport au placebo. Les données de l'une de ces études sont résumées dans le tableau 9.
Tableau 9: Paramètres glycémiques dans un essai contrôlé par placebo de 26 semaines
| Placebo N = 173 |
AVANDIA | AVANDIA | |||
| 4 mg une fois par jour N = 180 |
2 mg deux fois par jour N = 186 |
8 mg une fois par jour N = 181 |
4 mg deux fois par jour N = 187 |
||
| FPG (mg / dL) | |||||
| Référence (moyenne) | 225 | 229 | 225 | 228 | 228 |
| Changement par rapport à la référence (moyenne) | 8 | -25 | -35 | -42 | -55 |
| Différence par rapport au placebo (moyenne ajustée) | - | -31* | -43* | -49* | -62* |
| % de patients avec une diminution de ≤ 30 mg / dL par rapport à l'inclusion | 19% | 45% | 54% | 58% | 70% |
| HbA1c (%) | |||||
| Référence (moyenne) | 8.9 | 8.9 | 8.9 | 8.9 | 9.0 |
| Changement par rapport à la référence (moyenne) | 0.8 | 0.0 | -0.1 | -0.3 | -0.7 |
| Différence par rapport au placebo (moyenne ajustée) | - | -0.8* | -0.9* | -1.1* | -1.5* |
| % de patients avec une baisse de 0,7% de 0,7% par rapport à l'inclusion | 9% | 28% | 29% | 39% | 54% |
| * p <0,0001 par rapport au placebo. |
Lorsqu'il était administré à la même dose quotidienne totale, AVANDIA était généralement plus efficace pour réduire le FPG et l'HbA1c lorsqu'il était administré en doses divisées deux fois par jour par rapport à des doses une fois par jour. Cependant, pour l'HbA1c, la différence entre les doses de 4 mg une fois par jour et de 2 mg deux fois par jour n'était pas statistiquement significative.
Études cliniques à long terme
Le maintien à long terme de l'effet a été évalué dans un essai contrôlé par glyburide en double aveugle de 52 semaines chez des patients atteints de diabète de type 2. Les patients ont été randomisés pour recevoir AVANDIA 2 mg deux fois par jour (N = 195) ou AVANDIA 4 mg deux fois par jour (N = 189) ou du glyburide (N = 202) pendant 52 semaines. Les patients recevant du glyburide ont reçu une dose initiale de 2,5 mg / jour ou 5,0 mg / jour. La posologie a ensuite été titrée par incréments de 2,5 mg / jour au cours des 12 semaines suivantes, jusqu'à une posologie maximale de 15,0 mg / jour afin d'optimiser le contrôle glycémique. Par la suite, la dose de glyburide a été maintenue constante.
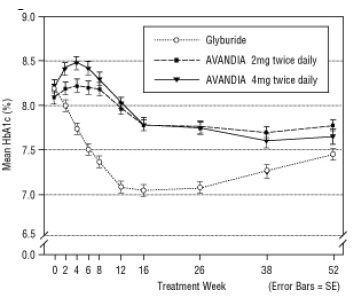
La dose médiane titrée de glyburide était de 7,5 mg. Tous les traitements ont entraîné une amélioration statistiquement significative du contrôle glycémique par rapport aux valeurs initiales (Figure 4 et Figure 5). À la fin de la semaine 52, la réduction du FPG et de l'HbA1c était de -40,8 mg / dL et de -0,53% avec AVANDIA 4 mg deux fois par jour; -25,4 mg / dL et -0,27% avec AVANDIA 2 mg deux fois par jour; et -30,0 mg / dL et -0,72% avec le glyburide. Pour l'HbA1c, la différence entre AVANDIA 4 mg deux fois par jour et le glyburide n'était pas statistiquement significative à la semaine 52. La chute initiale du FPG avec le glyburide a été plus importante qu'avec AVANDIA; cependant, cet effet était moins durable dans le temps.
L'amélioration du contrôle glycémique observée avec AVANDIA 4 mg deux fois par jour à la semaine 26 s'est maintenue jusqu'à la semaine 52 de l'étude.
Figure 4. FPG moyen dans le temps dans une étude contrôlée par le glyburide de 52 semaines
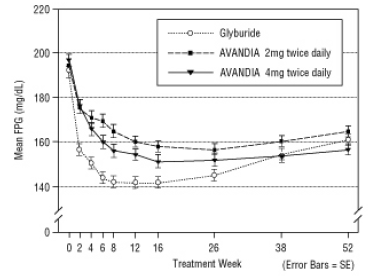
Figure 5. HbA1c moyenne au fil du temps dans une étude contrôlée par le glyburide de 52 semaines
Une hypoglycémie a été rapportée chez 12,1% des patients traités au glyburide contre 0,5% (2 mg deux fois par jour) et 1,6% (4 mg deux fois par jour) des patients traités par AVANDIA. Les améliorations du contrôle glycémique ont été associées à un gain de poids moyen de 1,75 kg et 2,95 kg pour patients traités respectivement avec 2 mg et 4 mg d'AVANDIA contre 1,9 kg chez les patients traités au glyburide les patients. Chez les patients traités par AVANDIA, les produits fractionnés C-peptide, insuline, pro-insuline et pro-insuline ont été significativement réduit de façon ordonnée par rapport à une augmentation de la dose de glyburide les patients.
Un essai de progression des résultats du diabète (ADOPT) était un essai multicentrique, en double aveugle, contrôlé (N = 4 351) mené sur 4 à 6 ans pour comparer l'innocuité et l'efficacité d'AVANDIA, de metformine et de glyburide en monothérapie chez des patients récemment diagnostiqués avec un diabète sucré de type 2 (¤ ¤ 3 ans) insuffisamment contrôlés par l'alimentation et exercice. L'âge moyen des patients dans cet essai était de 57 ans et la majorité des patients (83%) n'avaient aucun antécédent connu de maladie cardiovasculaire. Le FPG et l'HbA1c initiaux moyens étaient respectivement de 152 mg / dL et 7,4%. Les patients ont été randomisés pour recevoir soit 4 mg d'AVANDIA une fois par jour, 2,5 mg de glyburide une fois par jour, soit 500 mg de metformine une fois par jour, et les doses ont été titré pour un contrôle glycémique optimal jusqu'à un maximum de 4 mg deux fois par jour pour AVANDIA, 7,5 mg deux fois par jour pour le glyburide et 1000 mg deux fois par jour pour metformine. Le critère principal d'efficacité était le temps écoulé avant une FPG> 180 mg / dL consécutive après au moins 6 semaines de traitement au maximum dose tolérée du médicament à l'étude ou temps nécessaire à un contrôle glycémique inadéquat, tel que déterminé par une décision indépendante Comité.
L'incidence cumulée du critère principal d'efficacité à 5 ans était de 15% avec AVANDIA, 21% avec la metformine et 34% avec du glyburide (rapport de risque 0,68 [IC à 95% 0,55, 0,85] contre la metformine, HR 0,37 [IC à 95% 0,30, 0,45] contre le glyburide).
Les données cardiovasculaires et les événements indésirables (y compris les effets sur le poids corporel et les fractures osseuses) d'ADOPT pour AVANDIA, la metformine et le glyburide sont décrits dans AVERTISSEMENTS ET PRECAUTIONS et EFFETS INDÉSIRABLES, respectivement. Comme pour tous les médicaments, les résultats d'efficacité doivent être pris en compte avec les informations de sécurité pour évaluer le bénéfice et le risque potentiels pour un patient individuel.
Association avec la metformine ou la sulfonylurée
L'ajout d'AVANDIA à la metformine ou à la sulfonylurée a entraîné une réduction significative de l'hyperglycémie par rapport à l'un ou l'autre de ces agents seuls. Ces résultats sont cohérents avec un effet additif sur le contrôle glycémique lorsque AVANDIA est utilisé en association.
Association avec la metformine
Au total, 670 patients atteints de diabète de type 2 ont participé à deux essais randomisés en double aveugle de 26 semaines, études contrôlées versus placebo / actives conçues pour évaluer l'efficacité d'AVANDIA en association avec metformine. AVANDIA, administré selon des schémas posologiques une fois par jour ou deux fois par jour, a été ajouté à la thérapie des patients insuffisamment contrôlés à la dose maximale (2,5 grammes / jour) de metformine.
Dans une étude, des patients insuffisamment contrôlés avec 2,5 grammes / jour de metformine (moyenne initiale FPG 216 mg / dL et moyenne initiale HbA1c 8,8%) ont été randomisés pour recevoir 4 mg d'AVANDIA une fois par jour, 8 mg d'AVANDIA une fois par jour ou un placebo en plus de metformine. Une amélioration statistiquement significative de la FPG et de l'HbA1c a été observée chez les patients traités avec les combinaisons de metformine et 4 mg d'AVANDIA une fois par jour et 8 mg d'AVANDIA une fois par jour, par rapport aux patients qui ont continué avec la metformine seule (Tableau 10).
Tableau 10. Paramètres glycémiques dans une étude combinée de 26 semaines sur AVANDIA Plus Metformin
| Metformine N = 113 |
AVANDIA 4 mg une fois par jour + metformine N = 116 |
AVANDIA 8 mg une fois par jour + metformine N = 110 |
|
| FPG (mg / dL) | |||
| Référence (moyenne) | 214 | 215 | 220 |
| Changement par rapport à la référence (moyenne) | 6 | -33 | -48 |
| Différence par rapport à la metformine seule (moyenne ajustée) | - | -40* | -53* |
| % de patients avec une diminution de ≤ 30 mg / dL par rapport à l'inclusion | 20% | 45% | 61% |
| HbA1c (%) | |||
| Référence (moyenne) | 8.6 | 8.9 | 8.9 |
| Changement par rapport à la référence (moyenne) | 0.5 | -0.6 | -0.8 |
| Différence par rapport à la metformine seule (moyenne ajustée) | - | -1.0* | -1.2* |
| % de patients avec une baisse de 0,7% de 0,7% par rapport à l'inclusion | 11% | 45% | 52% |
| * p <0,0001 par rapport à la metformine. |
Dans une deuxième étude de 26 semaines, des patients atteints de diabète de type 2 mal contrôlés avec 2,5 grammes / jour de metformine ont été randomisés pour recevoir l'association d'AVANDIA 4 mg deux fois par jour et la metformine (N = 105) a montré une amélioration statistiquement significative du contrôle glycémique avec un effet de traitement moyen pour le FPG de -56 mg / dL et un effet de traitement moyen pour l'HbA1c de -0,8% par rapport à metformine seule. La combinaison de metformine et d'AVANDIA a entraîné des niveaux plus faibles de FPG et d'HbA1c que l'un ou l'autre agent seul.
Les patients insuffisamment contrôlés à la dose maximale (2,5 grammes / jour) de metformine et qui ont été passé en monothérapie avec AVANDIA a démontré une perte de contrôle glycémique, comme en témoignent les augmentations de FPG et HbA1c. Dans ce groupe, des augmentations des LDL et des VLDL ont également été observées.
Combinaison avec une sulfonylurée
Au total, 3 457 patients atteints de diabète de type 2 ont participé à dix études randomisées, en double aveugle, contrôlées versus placebo / actives de 24 à 26 semaines et une Étude en double aveugle de 2 ans, contrôlée par contrôle actif chez des patients âgés, conçue pour évaluer l'efficacité et la sécurité d'AVANDIA en association avec un sulfonylurée. AVANDIA 2 mg, 4 mg ou 8 mg par jour a été administré une fois par jour (3 études) ou en doses fractionnées deux fois par jour (7 études), aux patients insuffisamment contrôlés à une dose sous-maximale ou maximale de sulfonylurée.
Dans ces études, l'association d'AVANDIA à 4 mg ou à 8 mg par jour (administrée en doses divisées en une ou deux fois par jour) et sulfonylurée a réduit de manière significative la FPG et l'HbA1c par rapport au placebo plus sulfonylurée ou à une augmentation de la sulfonylurée. Le tableau 11 présente les données regroupées pour 8 études dans lesquelles AVANDIA ajouté à la sulfonylurée a été comparé au placebo plus sulfonylurée.
Tableau 11. Paramètres glycémiques dans les études combinées de 24 à 26 semaines d'AVANDIA Plus Sulfonylurée
| Dosage divisé deux fois par jour (5 études) | Sulfonylurée N = 397 |
AVANDIA 2 mg deux fois par jour + sulfonylurée N = 497 |
Sulfonylurée N = 248 |
AVANDIA 4 mg deux fois par jour + sulfonylurée N = 346 |
| FPG (mg / dL) | ||||
| Référence (moyenne) | 204 | 198 | 188 | 187 |
| Changement par rapport à la référence (moyenne) | 11 | -29 | 8 | -43 |
| Différence par rapport à la sulfonylurée seule (moyenne ajustée) | - | -42* | - | -53* |
| % de patients avec une diminution de ≤ 30 mg / dL par rapport à l'inclusion | 17% | 49% | 15% | 61% |
| HbA1c (%) | ||||
| Référence (moyenne) | 9.4 | 9.5 | 9.3 | 9.6 |
| Changement par rapport à la référence (moyenne) | 0.2 | -1.0 | 0.0 | -1.6 |
| Différence par rapport à la sulfonylurée seule (moyenne ajustée) | - | -1.1* | - | -1.4* |
| % de patients avec une baisse de 0,7% de 0,7% par rapport à l'inclusion | 21% | 60% | 23% | 75% |
| Dosage une fois par jour (3 études) | Sulfonylurée N = 172 |
AVANDIA 4 mg une fois par jour + sulfonylurée N = 172 |
Sulfonylurée N = 173 |
AVANDIA 8 mg une fois par jour + sulfonylurée N = 176 |
| FPG (mg / dL) | ||||
| Référence (moyenne) | 198 | 206 | 188 | 192 |
| Changement par rapport à la référence (moyenne) | 17 | -25 | 17 | -43 |
| Différence par rapport à la sulfonylurée seule (moyenne ajustée) | - | -47* | - | -66* |
| % de patients avec une diminution de ≤ 30 mg / dL par rapport à l'inclusion | 17% | 48% | 19% | 55% |
| HbA1c (%) | ||||
| Référence (moyenne) | 8.6 | 8.8 | 8.9 | 8.9 |
| Changement par rapport à la référence (moyenne) | 0.4 | -0.5 | 0.1 | -1.2 |
| Différence par rapport à la sulfonylurée seule (moyenne ajustée) | - | -0.9* | - | -1.4* |
| % de patients avec une baisse de 0,7% de 0,7% par rapport à l'inclusion | 11% | 36% | 20% | 68% |
| * p <0,0001 par rapport à la sulfonylurée seule. |
L'une des études de 24 à 26 semaines incluait des patients insuffisamment contrôlés aux doses maximales de glyburide et passés à 4 mg d'AVANDIA par jour en monothérapie; dans ce groupe, la perte de contrôle glycémique a été démontrée, comme en témoignent les augmentations du FPG et de l'HbA1c.
Dans une étude en double aveugle de 2 ans, des patients âgés (59 à 89 ans) sous sulfonylurée semi-maximale (glipizide 10 mg deux fois par jour) ont été randomisés pour l'ajout d'AVANDIA (n = 115, 4 mg une fois par jour à 8 mg au besoin) ou à une augmentation continue du glipizide (n = 110), jusqu'à un maximum de 20 mg deux fois du quotidien. La FPG et l'HbA1c initiales moyennes étaient respectivement de 157 mg / dL et 7,72% pour le bras AVANDIA plus glipizide et de 159 mg / dL et 7,65% pour le bras glipizide up-titration. Une perte de contrôle glycémique (FPG â ‰ ¥ 180 mg / dL) s'est produite chez une proportion significativement plus faible de patients (2%) sous AVANDIA plus glipizide par rapport aux patients du bras de sur-titration du glipizide (28,7%). Environ 78% des patients sous thérapie combinée ont terminé les 2 ans de thérapie tandis que seulement 51% ont terminé la monothérapie au glipizide. L'effet de la thérapie combinée sur le FPG et l'HbA1c a été durable au cours de la période d'étude de 2 ans, avec des patients atteignant une moyenne de 132 mg / dL pour le FPG et une moyenne de 6,98% pour l'HbA1c par rapport à aucun changement sur le glipizide bras.
Combinaison avec Sulfonylurea Plus Metformin
Dans deux études de 24 à 26 semaines, en double aveugle, contrôlées par placebo, conçues pour évaluer l'efficacité et l'innocuité d'AVANDIA en association avec sulfonylurée plus metformine, AVANDIA 4 mg ou 8 mg par jour, a été administré en doses divisées deux fois par jour à des patients insuffisamment contrôlés aux doses sous-maximales (10 mg) et maximales (20 mg) de glyburide et à la dose maximale de metformine (2 g / jour). Une amélioration statistiquement significative de la FPG et de l'HbA1c a été observée chez les patients traités par les combinaisons de sulfonylurée plus metformine et 4 mg d'AVANDIA et 8 mg d'AVANDIA versus patients ont continué sur sulfonylurée plus metformine, comme indiqué dans le tableau 12.
Tableau 12. Paramètres glycémiques dans une étude combinée de 26 semaines sur AVANDIA Plus Sulfonylurea et Metformin
| Sulfonylurée + metformine N = 273 |
AVANDIA 2 mg deux fois tous les jours + sulfonylurée + metformine N = 276 |
AVANDIA 4 mg deux fois par jour + sulfonylurée + metformine N = 277 |
|
| FPG (mg / dL) | |||
| Référence (moyenne) | 189 | 190 | 192 |
| Changement par rapport à la référence (moyenne) | 14 | -19 | -40 |
| Différence entre sulfonylurée et metformine (moyenne ajustée) | - | -30* | -52* |
| % de patients avec une diminution de ≤ 30 mg / dL par rapport à l'inclusion | 16% | 46% | 62% |
| HbA1c (%) | |||
| Référence (moyenne) | 8.7 | 8.6 | 8.7 |
| Changement par rapport à la référence (moyenne) | 0.2 | -0.4 | -0.9 |
| Différence entre sulfonylurée et metformine (moyenne ajustée) | - | -0.6* | -1.1* |
| % de patients avec une baisse de 0,7% de 0,7% par rapport à l'inclusion | 16% | 39% | 63% |
| * p <0,0001 par rapport au placebo. |
Haut
Les références
- Document d'information de la Food and Drug Administration. Réunion conjointe des comités consultatifs des médicaments endocriniens métaboliques et de la sécurité des médicaments et de la gestion des risques. Ju 2007.
- Enquêteurs du procès DREAM. Effet de la rosiglitazone sur la fréquence du diabète avec une tolérance au glucose altérée ou une glycémie à jeun altérée: un contrôle randomisé Lancette 2006;368:1096-1105.
- Accueil PD, Pocock SJ, Beck-Nielsen H, et al. Rosiglitazone évalué pour les résultats cardiovasculaires - une analyse intermédiaire. NEJM 2007; 357: 1-11.
- Park JY, Kim KA, Kang MH et al. Effet de la rifampicine sur la pharmacocinétique de la rosiglitazone chez des sujets sains. Clin Pharmacol Ther 2004; 75: 157-162.
Haut
Comment fournis / stockage et manutention
Chaque comprimé pentagonal pelliculé TILTAB contient de la rosiglitazone sous forme de maléate, comme suit: 2 mg de rose, en creux avec SB d'un côté et 2 de l'autre; 4 mg d'orange, en creux avec SB d'un côté et 4 de l'autre; 8 mg-brun-rouge, en creux avec SB d'un côté et 8 de l'autre.
- Flacons de 2 mg de 60: NDC 0029-3158-18
- Flacons de 4 mg de 30: NDC 0029-3159-13
- Flacons de 4 mg de 90: NDC 0029-3159-00
- Flacons de 8 mg de 30: NDC 0029-3160-13
- Flacons de 8 mg de 90: NDC 0029-3160-59
Conserver à 25 ° C (77 ° F); excursions 15 à 30 C (59 à 86 F). Distribuer dans un récipient étanche et résistant à la lumière.
dernière mise à jour 02/2008
Avandia, maléate de rosiglitazone, informations destinées aux patients (en anglais simple)
Informations détaillées sur les signes, les symptômes, les causes et les traitements du diabète
Les informations contenues dans cette monographie ne sont pas destinées à couvrir toutes les utilisations, directions, précautions, interactions médicamenteuses ou effets indésirables possibles. Ces informations sont généralisées et ne sont pas destinées à un avis médical spécifique. Si vous avez des questions sur les médicaments que vous prenez ou si vous souhaitez plus d'informations, consultez votre médecin, votre pharmacien ou votre infirmière.
retour à:Parcourir tous les médicaments pour le diabète


